Computational Design and Experimental Characterization of Metallopeptides As Proteases for Bioengineering Applications
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A-Ailable At
International Journal of Current Advanced Research ISSN: O: 2319-6475, ISSN: P: 2319 – 6505, Impact Factor: SJIF: 5.438 Available Online at www.journalijcar.org Volume 6; Issue 2; February 2017; Page No. 2131-2138 Research Article A COMPARATIVE ANALYSIS OF THERMOPHILIC AND PSYCHROPHILIC METALLOPEPTIDASES: INVOLVEMENT OF BIOINFORMATIC APPROACH Anuprabha1., AbhigyanNath2 and Radha Chaube3* 1,2 3Bioinformatic Division, MahilaMahavidyalya, Banaras Hindu University, Varanasi-221005, Uttar Pradesh, India ARTICLEDepartment INFO of Zoology, Institute of Science,ABSTRACT Banaras Hindu University, Varanasi-221005, Uttar Pradesh, India The thermolysin family of enzymes is classified as the M4 family of metallopeptidases. M4 Article History: family comprises numerous zinc-dependent metallopeptidases that hydrolyze peptide Received 9th November, 2016 bonds. Zinc containing peptidases are widely distributed in nature and have important roles Received in revised form 12thDecember, 2016 in many physiological processes. M4 family comprises numerous zinc-dependent Accepted 5th January, 2017 metallopeptidases that hydrolyse peptide bonds. Metallopeptidases were studied for the Published online 28th February, 2017 reason of their great relevance to biology, medicine and biotechnology. In the present study, detailed comparative analyses of both thermophilic and psychrophilic metallopeptidases were performed. The comparative analysis of both thermophilic and Key words: psychrophilic metallopeptidases was performed by taking sequence parameters such as Thermophilic, Psychrophilic, amino acid composition, amino acid property group composition, physico-chemical Metallopeptidases, amino acid residues, properties, secondary structure content. From comparison between the two groups, it was analysis found that Gln, Asn, Ser, Thr, and His are significantly lower in thermophilic metallopeptidase. Positively charged residues (Lys, Arg and Glu) are more significant in thermophilic metallopeptidases than in psychrophilic metallopeptidases. -

(12) United States Patent (10) Patent No.: US 6,395,889 B1 Robison (45) Date of Patent: May 28, 2002
USOO6395889B1 (12) United States Patent (10) Patent No.: US 6,395,889 B1 Robison (45) Date of Patent: May 28, 2002 (54) NUCLEIC ACID MOLECULES ENCODING WO WO-98/56804 A1 * 12/1998 ........... CO7H/21/02 HUMAN PROTEASE HOMOLOGS WO WO-99/0785.0 A1 * 2/1999 ... C12N/15/12 WO WO-99/37660 A1 * 7/1999 ........... CO7H/21/04 (75) Inventor: fish E. Robison, Wilmington, MA OTHER PUBLICATIONS Vazquez, F., et al., 1999, “METH-1, a human ortholog of (73) Assignee: Millennium Pharmaceuticals, Inc., ADAMTS-1, and METH-2 are members of a new family of Cambridge, MA (US) proteins with angio-inhibitory activity', The Journal of c: - 0 Biological Chemistry, vol. 274, No. 33, pp. 23349–23357.* (*) Notice: Subject to any disclaimer, the term of this Descriptors of Protease Classes in Prosite and Pfam Data patent is extended or adjusted under 35 bases. U.S.C. 154(b) by 0 days. * cited by examiner (21) Appl. No.: 09/392, 184 Primary Examiner Ponnathapu Achutamurthy (22) Filed: Sep. 9, 1999 ASSistant Examiner William W. Moore (51) Int. Cl." C12N 15/57; C12N 15/12; (74) Attorney, Agent, or Firm-Alston & Bird LLP C12N 9/64; C12N 15/79 (57) ABSTRACT (52) U.S. Cl. .................... 536/23.2; 536/23.5; 435/69.1; 435/252.3; 435/320.1 The invention relates to polynucleotides encoding newly (58) Field of Search ............................... 536,232,235. identified protease homologs. The invention also relates to 435/6, 226, 69.1, 252.3 the proteases. The invention further relates to methods using s s s/ - - -us the protease polypeptides and polynucleotides as a target for (56) References Cited diagnosis and treatment in protease-mediated disorders. -

The Extracellular Proteases Produced by Vibrio Parahaemolyticus
World Journal of Microbiology and Biotechnology (2018) 34:68 https://doi.org/10.1007/s11274-018-2453-4 REVIEW The extracellular proteases produced by Vibrio parahaemolyticus George Osei‑Adjei1 · Xinxiang Huang1 · Yiquan Zhang1 Received: 20 February 2018 / Accepted: 8 May 2018 © Springer Science+Business Media B.V., part of Springer Nature 2018 Abstract Vibrio parahaemolyticus, a Gram-negative bacterium, inhabits marine and estuarine environments and it is a major pathogen responsible globally for most cases of seafood-associated gastroenteritis in humans and acute hepatopancreatic necrosis syn- drome in shrimps. There has been a dramatic worldwide increase in V. parahaemolyticus infections over the last two decades. The pathogenicity of V. parahaemolyticus has been linked to the expression of different kinds of virulence factors including extracellular proteases, such as metalloproteases and serine proteases. V. parahaemolyticus expresses the metalloproteases; PrtV, VppC, VPM and the serine proteases; VPP1/Protease A, VpSP37, PrtA. Extracellular proteases have been identified as potential virulence factors which directly digest many kinds of host proteins or indirectly are involved in the processing of other toxic protein factors. This review summarizes findings on the metalloproteases and serine proteases produced by V. parahaemolyticus and their roles in infections. Identifying the role of V. parahaemolyticus virulence-associated extracellular proteases deepens our understanding of diseases caused by this bacterium. Keywords Extracellular protease · Metalloprotease · Serine protease · Vibrio parahaemolyticus · Virulence Introduction incubation period of V. parahaemolyticus is usually 12–24 h post-infection with infection usually occurring in the hot The Gram-negative bacterium, Vibrio parahaemolyticus, summer months because the bacteria is found free swim- lives in marine and estuarine environments and it is a major ming in coastal water or attached to fish and shellfish. -

Serine Proteases with Altered Sensitivity to Activity-Modulating
(19) & (11) EP 2 045 321 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 08.04.2009 Bulletin 2009/15 C12N 9/00 (2006.01) C12N 15/00 (2006.01) C12Q 1/37 (2006.01) (21) Application number: 09150549.5 (22) Date of filing: 26.05.2006 (84) Designated Contracting States: • Haupts, Ulrich AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 51519 Odenthal (DE) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Coco, Wayne SK TR 50737 Köln (DE) •Tebbe, Jan (30) Priority: 27.05.2005 EP 05104543 50733 Köln (DE) • Votsmeier, Christian (62) Document number(s) of the earlier application(s) in 50259 Pulheim (DE) accordance with Art. 76 EPC: • Scheidig, Andreas 06763303.2 / 1 883 696 50823 Köln (DE) (71) Applicant: Direvo Biotech AG (74) Representative: von Kreisler Selting Werner 50829 Köln (DE) Patentanwälte P.O. Box 10 22 41 (72) Inventors: 50462 Köln (DE) • Koltermann, André 82057 Icking (DE) Remarks: • Kettling, Ulrich This application was filed on 14-01-2009 as a 81477 München (DE) divisional application to the application mentioned under INID code 62. (54) Serine proteases with altered sensitivity to activity-modulating substances (57) The present invention provides variants of ser- screening of the library in the presence of one or several ine proteases of the S1 class with altered sensitivity to activity-modulating substances, selection of variants with one or more activity-modulating substances. A method altered sensitivity to one or several activity-modulating for the generation of such proteases is disclosed, com- substances and isolation of those polynucleotide se- prising the provision of a protease library encoding poly- quences that encode for the selected variants. -

Structural Basis of Mammalian Mucin Processing by the Human Gut O
ARTICLE https://doi.org/10.1038/s41467-020-18696-y OPEN Structural basis of mammalian mucin processing by the human gut O-glycopeptidase OgpA from Akkermansia muciniphila ✉ ✉ Beatriz Trastoy 1,4, Andreas Naegeli2,4, Itxaso Anso 1,4, Jonathan Sjögren 2 & Marcelo E. Guerin 1,3 Akkermansia muciniphila is a mucin-degrading bacterium commonly found in the human gut that promotes a beneficial effect on health, likely based on the regulation of mucus thickness 1234567890():,; and gut barrier integrity, but also on the modulation of the immune system. In this work, we focus in OgpA from A. muciniphila,anO-glycopeptidase that exclusively hydrolyzes the peptide bond N-terminal to serine or threonine residues substituted with an O-glycan. We determine the high-resolution X-ray crystal structures of the unliganded form of OgpA, the complex with the glycodrosocin O-glycopeptide substrate and its product, providing a comprehensive set of snapshots of the enzyme along the catalytic cycle. In combination with O-glycopeptide chemistry, enzyme kinetics, and computational methods we unveil the molecular mechanism of O-glycan recognition and specificity for OgpA. The data also con- tribute to understanding how A. muciniphila processes mucins in the gut, as well as analysis of post-translational O-glycosylation events in proteins. 1 Structural Biology Unit, Center for Cooperative Research in Biosciences (CIC bioGUNE), Basque Research and Technology Alliance (BRTA), Bizkaia Technology Park, Building 801A, 48160 Derio, Spain. 2 Genovis AB, Box 790, 22007 Lund, Sweden. 3 IKERBASQUE, Basque Foundation for Science, 48013 ✉ Bilbao, Spain. 4These authors contributed equally: Beatriz Trastoy, Andreas Naegeli, Itxaso Anso. -
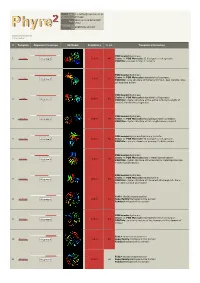
Phyre 2 Results for P22894
Email [email protected] Description P22894 Wed Jun 6 09:54:50 BST Date 2012 Unique Job eba6f50a2e10c91f ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:hydrolase 1 c1gxdA_ Alignment 100.0 46 Chain: A: PDB Molecule:72 kda type iv collagenase; PDBTitle: prommp-2/timp-2 complex PDB header:hydrolase Chain: A: PDB Molecule:interstitial collagenase; 2 c1su3A_ 100.0 61 Alignment PDBTitle: x-ray structure of human prommp-1: new insights into2 collagenase action PDB header:hydrolase Chain: B: PDB Molecule:interstitial collagenase; 3 c2cltB_ 100.0 61 Alignment PDBTitle: crystal structure of the active form (full-length) of human2 fibroblast collagenase. PDB header:hydrolase 4 c3ba0A_ Alignment 100.0 49 Chain: A: PDB Molecule:macrophage metalloelastase; PDBTitle: crystal structure of full-length human mmp-12 PDB header:hydrolase/hydrolase inhibitor 5 c1eakA_ Alignment 100.0 52 Chain: A: PDB Molecule:72 kda type iv collagenase; PDBTitle: catalytic domain of prommp-2 e404q mutant PDB header:hydrolase Chain: A: PDB Molecule:matrix metalloproteinase-9; 6 c1l6jA_ 100.0 56 Alignment PDBTitle: crystal structure of human matrix metalloproteinase mmp92 (gelatinase b). PDB header:hydrolase Chain: A: PDB Molecule:stromelysin-1; 7 c1slmA_ 100.0 54 Alignment PDBTitle: crystal structure of fibroblast stromelysin-1: the c- truncated human2 proenzyme Fold:4-bladed beta-propeller 8 d1fbla1 Alignment 100.0 57 Superfamily:Hemopexin-like domain Family:Hemopexin-like domain PDB header:hydrolase -

Structural Interaction of Natural and Synthetic Inhibitors with the Venom Metalloproteinase, Atrolysin C
Proc. Nati. Acad. Sci. USA Vol. 91, pp. 8447-8451, August 1994 Biochemistry Structural interaction of natural and synthetic inhibitors with the venom metalloproteinase, atrolysin C (form d) (coilagenase/inhibltor complex/crystafography/metastasis) DACHUAN ZHANG*, ISTVAN BOTOS*, FRANZ-XAVER GOMIS-ROTHtt, RONALD DOLL§, CHRISTINE BLOOD§, F. GEORGE NJOROGE§, JAY W. Fox¶, WOLFRAM BODEt, AND EDGAR F. MEYER* 11 *Biographics Laboratory, Department of Biochemistry and Biophysics, Texas A&M University, College Station, TX 77843; tMax Planck Institute of Biochemistry, D-82152 Martinsried, Germany; §Schering-Plough Research Institute, 2015 Galloping Hill Road, Kenilworth, NJ 07033; IBiomolecular Research Facility, University of Virginia Health Sciences Center, Charlottesville, VA 22908 Communicated by Derek H. R. Barton, May 20, 1994 ABSTRACT The structure of the metalloproteinase and 80% sequence similarity to Ht-d) has been described (12), as hemorrhagic toxin atrolysin C form d (EC 3.4.24.42), from the was the native digestive MP, astacin (13). A high degree of venom ofthe western diamondback rattlesnake Crotalus atrox, tertiary structure conservation among the astacin, reprol- has been determined to atomic resolution by x-ray crystallo- ysin, serralysins, and the MMP subfamilies is observed (9, 14) graphic methods. This study illuminates the nature ofinhibitor at the active site, suggesting that the structural principles that binding with natural (<Glu-Asn-Trp, where <Glu is pyroglu- govern the interaction of substrates and inhibitors with tamic acid) and synthetic (SCH 47890) ligands. The primary members of these subfamilies are likely to be similar if not specificity pocket is exceptionally deep; the nature of inhibitor identical. Substrates and synthetic inhibitors of the MMPs and productive substrate binding is discussed. -

Genomic and Proteomic Analyses of the Coral Pathogen Vibrio Coralliilyticus Reveal a Diverse Virulence Repertoire
The ISME Journal (2011) 5, 1471–1483 & 2011 International Society for Microbial Ecology All rights reserved 1751-7362/11 www.nature.com/ismej ORIGINAL ARTICLE Genomic and proteomic analyses of the coral pathogen Vibrio coralliilyticus reveal a diverse virulence repertoire Eidy de O Santos1, Nelson Alves Jr1, Graciela M Dias1, Ana Maria Mazotto2, Alane Vermelho2, Gary J Vora3, Bryan Wilson4, Victor H Beltran4, David G Bourne4, Fre´de´rique Le Roux5,6 and Fabiano L Thompson1 1Laboratory of Microbiology, Institute of Biology, Federal University of Rio de Janeiro (UFRJ), Rio de Janeiro, Brazil; 2Laboratory of Proteases, Institute of Microbiology, UFRJ, Rio de Janeiro, Brazil; 3Center for Bio/ Molecular Science & Engineering, Naval Research Laboratory, Washington, DC, USA; 4Centre for Marine Microbiology and Genetics, Australian Institute of Marine Science, Townsville MC, Queensland, Australia; 5FR2424 CNRS UPMC Station Biologique de Roscoff, France and 6Ifremer, Laboratoire de Physiologie des Inverte´bre´s, Brest, France Vibrio coralliilyticus has been implicated as an important pathogen of coral species worldwide. In this study, the nearly complete genome of Vibrio coralliilyticus strain P1 (LMG23696) was sequenced and proteases implicated in virulence of the strain were specifically investigated. The genome sequence of P1 (5 513 256 bp in size) consisted of 5222 coding sequences and 58 RNA genes (53 tRNAs and at least 5 rRNAs). Seventeen metalloprotease and effector (vgrG, hlyA and hcp) genes were identified in the genome and expressed proteases were also detected in the secretome of P1. As the VcpA zinc-metalloprotease has been considered an important virulence factor of V. coralliilyticus, a vcpA deletion mutant was constructed to evaluate the effect of this gene in animal pathogenesis. -

Composition Containing Protease Produced by Vibrio and Method of Use in Debridement and Wound Healing
Europaisches Patentamt European Patent Office 0 Publication number: 0 472 011 A1 Office europeen des brevets EUROPEAN PATENT APPLICATION 0 Application number: 91112732.2 int. CI 5: A61K 37/48, C12N 9/52, C12N 15/31, //(C12N 15/31, 0 Date of filing: 29.07.91 C12R1:63) 0 Priority: 15.08.90 US 567884 0 Inventor: Fortney, Donald Zane 13.03.91 US 670612 2114 Reese Road Westminster, Md. 21157(US) 0 Date of publication of application: Inventor: Durham, Donald Richard 26.02.92 Bulletin 92/09 20617 Beaver Ridge Road Gaithersburg, Md. 20879(US) 0 Designated Contracting States: AT BE CH DE DK ES FR GB GR IT LI LU NL SE 0 Representative: UEXKULL & STOLBERG 0 Applicant: W.R. Grace & Co.-Conn. Patentanwalte Grace Plaza, 1114 Ave. of the Americas Beselerstrasse 4 New York New York 10036(US) W-2000 Hamburg 52(DE) 0 Composition containing protease produced by vibrio and method of use in debridement and wound healing. 0 A composition for treating wounds comprising at least one pharmaceutically acceptable carrier admixed with an effective amount of a protease selected from the group consisting of: (a) an extracellular neutral protease produced by cultivation of a microorganism belonging to the genus Vibrio, said protease characterized by the following properties: i. hydrolyzes components of necrotic tissue including denatured collagen, elastin and fribrin; ii. does not substantially hydrolyze native tissue in viva, and iii. exhibits stable activity when stored at 25° C in a topical formulation; (b) a protease expressed by recombinant host cells which have been transformed or transfected with an expression vector for said protease (a); and (c) mutants and hybrids of proteases (a) and (b) which are characterized by the properties (i) to (iii). -

Handbook of Proteolytic Enzymes Second Edition Volume 1 Aspartic and Metallo Peptidases
Handbook of Proteolytic Enzymes Second Edition Volume 1 Aspartic and Metallo Peptidases Alan J. Barrett Neil D. Rawlings J. Fred Woessner Editor biographies xxi Contributors xxiii Preface xxxi Introduction ' Abbreviations xxxvii ASPARTIC PEPTIDASES Introduction 1 Aspartic peptidases and their clans 3 2 Catalytic pathway of aspartic peptidases 12 Clan AA Family Al 3 Pepsin A 19 4 Pepsin B 28 5 Chymosin 29 6 Cathepsin E 33 7 Gastricsin 38 8 Cathepsin D 43 9 Napsin A 52 10 Renin 54 11 Mouse submandibular renin 62 12 Memapsin 1 64 13 Memapsin 2 66 14 Plasmepsins 70 15 Plasmepsin II 73 16 Tick heme-binding aspartic proteinase 76 17 Phytepsin 77 18 Nepenthesin 85 19 Saccharopepsin 87 20 Neurosporapepsin 90 21 Acrocylindropepsin 9 1 22 Aspergillopepsin I 92 23 Penicillopepsin 99 24 Endothiapepsin 104 25 Rhizopuspepsin 108 26 Mucorpepsin 11 1 27 Polyporopepsin 113 28 Candidapepsin 115 29 Candiparapsin 120 30 Canditropsin 123 31 Syncephapepsin 125 32 Barrierpepsin 126 33 Yapsin 1 128 34 Yapsin 2 132 35 Yapsin A 133 36 Pregnancy-associated glycoproteins 135 37 Pepsin F 137 38 Rhodotorulapepsin 139 39 Cladosporopepsin 140 40 Pycnoporopepsin 141 Family A2 and others 41 Human immunodeficiency virus 1 retropepsin 144 42 Human immunodeficiency virus 2 retropepsin 154 43 Simian immunodeficiency virus retropepsin 158 44 Equine infectious anemia virus retropepsin 160 45 Rous sarcoma virus retropepsin and avian myeloblastosis virus retropepsin 163 46 Human T-cell leukemia virus type I (HTLV-I) retropepsin 166 47 Bovine leukemia virus retropepsin 169 48 -

Structural, Functional and Therapeutic Aspects of Snake Venom Metal- Loproteinases
Send Orders for Reprints to [email protected] 28 Mini-Reviews in Organic Chemistry, 2014, 11, 28-44 Structural, Functional and Therapeutic Aspects of Snake Venom Metal- loproteinases P. Chellapandi* Department of Bioinformatics, School of Life Sciences, Bharathidasan University, Tiruchirappalli-620024, Tamil Nadu, India Abstract: Snake venoms are rich sources of metalloproteinases that are of biological interest due to their diverse molecu- lar diversity and selective therapeutic applications. Snake venoms metalloproteinases (SVMPs) belong to the MEROPS peptidase family M12B or reprolysin subfamily, which are consisted of four major domains include a reprolysin catalytic domain, a disintegrin domain, a reprolysin family propeptide domain and a cysteine-rich domain. The appropriate struc- tural and massive sequences information have been available for SVMPs family of enzymes in the Protein Data Bank and National Center for Biotechnology Information, respectively. Functional essentiality of every domain and a crucial contri- bution of binding geometry, primary specificity site, and structural motifs have been studied in details, pointing the way for designing potential anti-coagulation, antitumor, anti-complementary and anti-inflammatory drugs or peptides. These enzymes have been reported to degrade fibrinogen, fibrin and collagens, and to prevent progression of clot formation. An- giotensin-converting enzyme activity, antibacterial properties, haemorrhagic activity and platelet aggregation response of SVMPs have been studied earlier. Structural information of these enzymes together with recombinant DNA technology would strongly promote the construction of many recombinant therapeutic peptides, particularly fibrinogenases and vac- cines. We have comprehensively reviewed the structure-function-evolution relationships of SVMPs family proteins and their advances in the promising target models for structure-based inhibitors and peptides design. -

Genetic Basis for Variation of Metalloproteinase-Associated Biochemical Activity in Venom of the Mojave Rattlesnake (Crotalus Scutulatus Scutulatus)
Hindawi Publishing Corporation Biochemistry Research International Volume 2013, Article ID 251474, 11 pages http://dx.doi.org/10.1155/2013/251474 Research Article Genetic Basis for Variation of Metalloproteinase-Associated Biochemical Activity in Venom of the Mojave Rattlesnake (Crotalus scutulatus scutulatus) Ruben K. Dagda,1 Sardar Gasanov,2 Ysidro De La OIII,3 Eppie D. Rael,3 and Carl S. Lieb3 1 Pharmacology Department, University of Nevada School of Medicine, Manville Building 19A, Reno, NV 89557, USA 2 Science Department, Tashkent Ulugbek International School, 5-A J. Shoshiy Street, 100100 Tashkent, Uzbekistan 3 Department of Biological Sciences, University of Texas at El Paso, 500 West University Avenue, El Paso, TX 79968, USA Correspondence should be addressed to Ruben K. Dagda; [email protected] Received 26 April 2013; Accepted 25 June 2013 AcademicEditor:R.J.Linhardt Copyright © 2013 Ruben K. Dagda et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The metalloproteinase composition and biochemical profiles of rattlesnake venom can be highly variable among rattlesnakes ofthe same species. We have previously shown that the neurotoxic properties of the Mojave rattlesnake (Crotalus scutulatus scutulatus) are associated with the presence of the Mojave toxin A subunit suggesting the existence of a genetic basis for rattlesnake venom composition. In this report, we hypothesized the existence of a genetic basis for intraspecies variation in metalloproteinase- associated biochemical properties of rattlesnake venom of the Mojave rattlesnake. To address this question, we PCR-amplified and compared the genomic DNA nucleotide sequences that code for the mature metalloproteinase domain of fourteen Mojave rattlesnakes captured from different geographical locations across the southwest region of the United States.