Arxiv:1406.3917V1 [Cond-Mat.Mtrl-Sci]
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

Primary-Explosives
Improvised Primary Explosives © 1998 Dirk Goldmann No part of the added copyrighted parts (except brief passages that a reviewer may quote in a review) may be reproduced in any form unless the reproduced material includes the following two sentences: The copyrighted material may be reproduced without obtaining permission from anyone, provided: (1) all copyrighted material is reproduced full-scale. WARNING! Explosives are danegerous. In most countries it's forbidden to make them. Use your mind. You as an explosives expert should know that. 2 CONTENTS Primary Explosives ACETONE PEROXIDE 4 DDNP/DINOL 6 DOUBLE SALTS 7 HMTD 9 LEAD AZIDE 11 LEAD PICRATE 13 MEKAP 14 MERCURY FULMINATE 15 "MILK BOOSTER" 16 NITROMANNITE 17 SODIUM AZIDE 19 TACC 20 Exotic and Friction Primers LEAD NITROANILATE 22 NITROGEN SULFIDE 24 NITROSOGUANIDINE 25 TETRACENE 27 CHLORATE-FRICTION PRIMERS 28 CHLORATE-TRIMERCURY-ACETYLIDE 29 TRIHYDRAZINE-ZINC (II) NITRATE 29 Fun and Touch Explosives CHLORATE IMPACT EXPLOSIVES 31 COPPER ACETYLIDE 32 DIAMMINESILVER II CHLORATE 33 FULMINATING COPPER 33 FULMINATING GOLD 34 FULMINATING MERCURY 35 FULMINATING SILVER 35 NITROGEN TRICHLORIDE 36 NITROGEN TRIIODIDE 37 SILVER ACETYLIDE 38 SILVER FULMINATE 38 "YELLOW POWDER" 40 Latest Additions 41 End 3 PRIMARY EXPLOSIVES ACETONE PEROXIDE Synonyms: tricycloacetone peroxide, acetontriperoxide, peroxyacetone, acetone hydrogen explosive FORMULA: C9H18O6 VoD: 3570 m/s @ 0.92 g/cc. 5300 m/s @ 1.18 g/cc. EQUIVALENCE: 1 gram = No. 8 cap .75 g. = No. 6 cap SENSITIVITY: Very sensitive to friction, flame and shock; burns violently and can detonate even in small amounts when dry. DRAWBACKS: in 10 days at room temp. 50 % sublimates; it is best made immediately before use. -

Chemical List
1 EXHIBIT 1 2 CHEMICAL CLASSIFICATION LIST 3 4 1. Pyrophoric Chemicals 5 1.1. Aluminum alkyls: R3Al, R2AlCl, RAlCl2 6 Examples: Et3Al, Et2AlCl, EtAlCl2, Me3Al, Diethylethoxyaluminium 7 1.2. Grignard Reagents: RMgX (R=alkyl, aryl, vinyl X=halogen) 8 1.3. Lithium Reagents: RLi (R = alkyls, aryls, vinyls) 9 Examples: Butyllithium, Isobutyllithium, sec-Butyllithium, tert-Butyllithium, 10 Ethyllithium, Isopropyllithium, Methyllithium, (Trimethylsilyl)methyllithium, 11 Phenyllithium, 2-Thienyllithium, Vinyllithium, Lithium acetylide ethylenediamine 12 complex, Lithium (trimethylsilyl)acetylide, Lithium phenylacetylide 13 1.4. Zinc Alkyl Reagents: RZnX, R2Zn 14 Examples: Et2Zn 15 1.5. Metal carbonyls: Lithium carbonyl, Nickel tetracarbonyl, Dicobalt octacarbonyl 16 1.6. Metal powders (finely divided): Bismuth, Calcium, Cobalt, Hafnium, Iron, 17 Magnesium, Titanium, Uranium, Zinc, Zirconium 18 1.7. Low Valent Metals: Titanium dichloride 19 1.8. Metal hydrides: Potassium Hydride, Sodium hydride, Lithium Aluminum Hydride, 20 Diethylaluminium hydride, Diisobutylaluminum hydride 21 1.9. Nonmetal hydrides: Arsine, Boranes, Diethylarsine, diethylphosphine, Germane, 22 Phosphine, phenylphosphine, Silane, Methanetellurol (CH3TeH) 23 1.10. Non-metal alkyls: R3B, R3P, R3As; Tributylphosphine, Dichloro(methyl)silane 24 1.11. Used hydrogenation catalysts: Raney nickel, Palladium, Platinum 25 1.12. Activated Copper fuel cell catalysts, e.g. Cu/ZnO/Al2O3 26 1.13. Finely Divided Sulfides: Iron Sulfides (FeS, FeS2, Fe3S4), and Potassium Sulfide 27 (K2S) 28 REFERRAL -

Prohibited and Restricted Chemical List
School Emergency Response Plan and Management Guide Prohibited and Restricted Chemical List PROHIBITED AND RESTRICTED CHEMICAL LIST Introduction After incidents of laboratory chemical contamination at several schools, DCPS, The American Association for the Advancement of Science (AAAS) and DC Fire and Emergency Management Services developed an aggressive program for chemical control to eliminate student and staff exposure to potential hazardous chemicals. Based upon this program, all principals are required to conduct a complete yearly inventory of all chemicals located at each school building to identify for the removal and disposal of any prohibited/banned chemicals. Prohibited chemicals are those that pose an inherent, immediate, and potentially life- threatening risk, injury, or impairment due to toxicity or other chemical properties to students, staff, or other occupants of the school. These chemicals are prohibited from use and/or storage at the school, and the school is prohibited from purchasing or accepting donations of such chemicals. Restricted chemicals are chemicals that are restricted by use and/or quantities. If restricted chemicals are present at the school, each storage location must be addressed in the school's written emergency plan. Also, plan maps must clearly denote the storage locations of these chemicals. Restricted chemicals—demonstration use only are a subclass in the Restricted chemicals list that are limited to instructor demonstration. Students may not participate in handling or preparation of restricted chemicals as part of a demonstration. If Restricted chemicals—demonstration use only are present at the school, each storage location must be addressed in the school's written emergency plan. Section 7: Appendices – October 2009 37 School Emergency Response Plan and Management Guide Prohibited and Restricted Chemical List Following is a table of chemicals that are Prohibited—banned, Restricted—academic curriculum use, and Restricted—demonstration use only. -

United States Patent Office Patented Mar
3,311,468 United States Patent Office Patented Mar. 28, 1967 2 3,351,468 continuously or batchwise as desired. Other objects and SVRESR RECGVERY PRO CESS advantages will appear as the description proceeds. Charies Davidoff, 1:8 Roliagiiii Road, The attainment of the above objects is made possible Maiaasset, N.Y. 1936 by my method of recovering silver from a solution of No Drawing, Fied Dec. 26, 1963, Ser. No. 333,635 5 alkali metal silver cyanide comprising adding to the solu 6 Caias. (Ci. 75-63) tion at least about 0.25 mole of an alkali metal hydro sulfite compound for each mole of silver in the solution, This invention relates to a method for recovering silver, maintaining the resulting solution, preferably at an ele and more particularly to a method for recovering silver vated temperature above about 150 F., until cessation from a solution of alkali metal silver cyanide or other sil 10 of precipitation of the silver, and removing the precipi wer salt. tated silver from the solution. The above process has It is common knowledge that solutions of silver cyanide been found to constitute a surprisingly simple, economical complex exist in several fields of industry from which the method of expeditiously recovering silver from solutions silver must be recovered for obvious reasons of economy. of alkali metal silver cyanide in up to 100% yields with Thus, in the mining industry, a widely employed method out the need for the previously required acid-proof equip of recovering silver from ore containing the same involves ment. Further, although the said process exhibits its leaching the silver from the ore with an aqueous solu greatest advantages in the treatment of solutions contain tion of alkali metal cyanide, a solution of sodium or ing the highly stable alkali metal silver cyanide complex, potassium silver cyanide being thereby obtained. -

List of Reactive Chemicals
LIST OF REACTIVE CHEMICALS Chemical Prefix Chemical Name Reactive Reactive Reactive CAS# Chemical Chemical Chemical Stimulus 1 Stimulus 2 Stimulus 3 111-90-0 "CARBITOL" SOLVENT D 111-15-9 "CELLOSOLVE" ACETATE D 110-80-5 "CELLOSOLVE" SOLVENT D 2- (2,4,6-TRINITROPHENYL)ETHYL ACETATE (1% IN ACETONE & BENZENE S 12427-38-2 AAMANGAN W 88-85-7 AATOX S 40487-42-1 AC 92553 S 105-57-7 ACETAL D 75-07-0 ACETALDEHYDE D 105-57-7 ACETALDEHYDE, DIETHYL ACETAL D 108-05-4 ACETIC ACID ETHENYL ESTER D 108-05-4 ACETIC ACID VINYL ESTER D 75-07-0 ACETIC ALDEHYDE D 101-25-7 ACETO DNPT T 126-84-1 ACETONE DIETHYL ACETAL D 108-05-4 ACETOXYETHYLENE D 108-05-4 1- ACETOXYETHYLENE D 37187-22-7 ACETYL ACETONE PEROXIDE, <=32% AS A PASTE T 37187-22-7 ACETYL ACETONE PEROXIDE, <=42% T 37187-22-7 ACETYL ACETONE PEROXIDE, >42% T S 644-31-5 ACETYL BENZOYL PEROXIDE (SOLID OR MORE THAN 45% IN SOLUTION) T S 644-31-5 ACETYL BENZOYL PEROXIDE, <=45% T 506-96-7 ACETYL BROMIDE W 75-36-5 ACETYL CHLORIDE W ACETYL CYCLOHEXANE SULFONYL PEROXIDE (>82% WITH <12% WATER) T S 3179-56-4 ACETYL CYCLOHEXANE SULFONYL PEROXIDE, <=32% T 3179-56-4 ACETYL CYCLOHEXANE SULFONYL PEROXIDE, <=82% T 674-82-8 ACETYL KETENE (POISON INHALATION HAZARD) D 110-22-5 ACETYL PEROXIDE, <=27% T 110-22-5 ACETYL PEROXIDE, SOLID, OR MORE THAN 27% IN SOLUTION T S 927-86-6 ACETYLCHOLINE PERCHLORATE O S 74-86-2 ACETYLENE D 74-86-2 ACETYLENE (LIQUID) D ACETYLENE SILVER NITRATE D 107-02-08 ACRALDEHYDE (POISON INHALATION HAZARD) D 79-10-7 ACROLEIC ACID D 107-02-08 ACROLEIN, INHIBITED (POISON INHALATION HAZARD) D 107-02-08 ACRYLALDEHYDE (POISON INHALATION HAZARD) D 79-10-7 ACRYLIC ACID D 141-32-2 ACRYLIC ACID BUTYL ESTER D 140-88-5 ACRYLIC ACID ETHYL ESTER D 96-33-3 ACRYLIC ACID METHYL ESTER D Stimulus - Stimuli is the thermal, physical or chemical input needed to induce a hazardous reaction. -

The First Chemical Achievements and Publications by Justus Von Liebig
MICROREVIEW The First Chemical Achievements and Publications by Justus von Liebig 1803؊1873) on Metal Fulminates and Some Further Developments in Metal) Fulminates and Related Areas of Chemistry[‡] Wolfgang Beck[a] Dedicated to Professor Wolfgang Steglich on the occasion of his 70th birthday for his great friendship, collaboration and support[‡‡] Keywords: Liebig / Fulminates / Silver / Structure elucidation The first chemical investigations and publications by Justus by Liebig. Later, explosive (fulminato)metal complexes were von Liebig dealt with the fulminates of silver and mercury. prepared by Nef, Wieland, and especially by Lothar Wöhler Even as a boy Liebig had learnt how to prepare silver fulmi- and co-workers. In Munich the HCNO structure of fulminic nate, and as student of chemistry in Erlangen (1821) he stud- acid was established by its IR spectrum and the spectroscopic ied the properties and reactions of silver fulminate. In Paris in properties of (fulminato)metal complexes were studied. A se- 1823 (together with Gay-Lussac) he succeeded in analyzing ries of new nonexplosive complexes could be obtained by quantitatively the highly explosive silver compound. This dilution of the energy-rich species with large cations or li- great experimental success with the dangerous silver fulmi- gands. Recent X-ray structure determinations have revealed nate was most important in three respects: i. The develop- the almost perfect linear, tetrahedral, square-planar, or octa- ment of the experimental method later culminated in Liebig’s hedral structures of these complexes with linear − 2− perfected and well-known C,H,N analysis of organic com- metal−CϵNO bonds, e.g. [Au(CNO)2] , [Zn(CNO)4] , 2− 3− pounds (1830). -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -
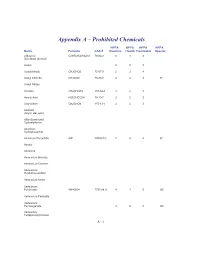
Appendix a – Prohibited Chemicals
Appendix A – Prohibited Chemicals NFPA NFPA NFPA NFPA Name Formula CAS #Reactive Health Flammable Special 2-Butanol C2H5CH(OH)CH3 78-92-2 0 1 3 (Sec-Butyl Alcohol) Acetal 023 Acetaldehyde CH3CHO5 75-07-0 2 3 4 Acetyl Chloride CH3COCl 75-36-5 2 3 3 W Acetyl Nitrate Acrolein CH2CHCHO 107-02-8 3 4 3 Acrylic Acid H2CCHCO2H 79-10-7 2 2 2 Acrylonitrile CH2CHCN 107-13-1 2 4 3 Alcohols (Allylic, Benzylic) Alkly-Substituted Cycloaliphatics Aluminum Hydrophosphide Aluminum Phosphide AlP 20859-73- 2 4 4 W Amatol Ammonal Ammonium Bromate Ammonium Chlorate Ammonium Hexanitrocobaltate Ammonium Nitrite Ammonium Perchlorate NH4ClO4 7790-98-9 4 1 0 OX Ammonium Periodate Ammonium Permanganate 300 OX Ammonium Tetraperoxychromate A - 1 Appendix A – Prohibited Chemicals NFPA NFPA NFPA NFPA Name Formula CAS #Reactive Health Flammable Special Antimony Compounds Arsenic And Arsenic Compounds Azides Azidocarbonyl Guanidine Barium Ba 2 2 1 W Barium Chlorate Ba(ClO3)2*H2O 13477-00- 1 2 0 OX Barium Oxide (Anhydrous) BaO 1304-28-5 2 3 0 Barium Peroxide BaO2 1304-29-6 0 1 0 OX Benzene C6H6 71-43-2 0 2 3 Benzene Diazonium Chloride Benzotriazole C6H5N3 95-14-7 0 2 1 Benzoyl Peroxide (C6H5CO)2O2 94-36-0 4 1 4 OX Benzyl Alcohol C6H5CH2OH 100-51-6 0 2 1 Bismuth Nitrate Bi(NO3)3*5H2O 10035-06- 3 1 0 OX Borane,Boranes, Diboranes Boron Tribromide 230 W Boron Trifluoride 140 Bromine Pentafluoride Brf5 7789-30-2 3 4 0 W,O Bromine Trifluoride 3 4 0 W,O Butadiene C4H6/CH2=(CH)2=CH 106-99-0 0 2 4 Butenetroil Trinitrate Cadmium and Cadmium Compounds Calcium Nitrate, Anhydrous Ca(NO3)2 -

Commerce in Explosives; 2020 Annual Those on the Annual List
Federal Register / Vol. 85, No. 247 / Wednesday, December 23, 2020 / Notices 83999 inspection at the Office of the Secretary or synonyms in brackets. This list Black powder substitutes. and on EDIS.3 supersedes the List of Explosive *Blasting agents, nitro-carbo-nitrates, This action is taken under the Materials published in the Federal including non-cap sensitive slurry and authority of section 337 of the Tariff Act Register on January 2, 2020 (Docket No. water gel explosives. of 1930, as amended (19 U.S.C. 1337), 2019R–04, 85 FR 128). Blasting caps. and of §§ 201.10 and 210.8(c) of the The 2020 List of Explosive Materials Blasting gelatin. Commission’s Rules of Practice and is a comprehensive list, but is not all- Blasting powder. Procedure (19 CFR 201.10, 210.8(c)). inclusive. The definition of ‘‘explosive BTNEC [bis (trinitroethyl) carbonate]. materials’’ includes ‘‘[e]xplosives, BTNEN [bis (trinitroethyl) nitramine]. By order of the Commission. BTTN [1,2,4 butanetriol trinitrate]. Issued: December 18, 2020. blasting agents, water gels and detonators. Explosive materials, Bulk salutes. William Bishop, include, but are not limited to, all items Butyl tetryl. Supervisory Hearings and Information in the ‘List of Explosive Materials’ Officer. C provided for in § 555.23.’’ 27 CFR Calcium nitrate explosive mixture. [FR Doc. 2020–28458 Filed 12–22–20; 8:45 am] 555.11. Accordingly, the fact that an BILLING CODE 7020–02–P Cellulose hexanitrate explosive explosive material is not on the annual mixture. list does not mean that it is not within Chlorate explosive mixtures. coverage of the law if it otherwise meets DEPARTMENT OF JUSTICE Composition A and variations. -

The Elements.Pdf
A Periodic Table of the Elements at Los Alamos National Laboratory Los Alamos National Laboratory's Chemistry Division Presents Periodic Table of the Elements A Resource for Elementary, Middle School, and High School Students Click an element for more information: Group** Period 1 18 IA VIIIA 1A 8A 1 2 13 14 15 16 17 2 1 H IIA IIIA IVA VA VIAVIIA He 1.008 2A 3A 4A 5A 6A 7A 4.003 3 4 5 6 7 8 9 10 2 Li Be B C N O F Ne 6.941 9.012 10.81 12.01 14.01 16.00 19.00 20.18 11 12 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 3 Na Mg IIIB IVB VB VIB VIIB ------- VIII IB IIB Al Si P S Cl Ar 22.99 24.31 3B 4B 5B 6B 7B ------- 1B 2B 26.98 28.09 30.97 32.07 35.45 39.95 ------- 8 ------- 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 4 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 39.10 40.08 44.96 47.88 50.94 52.00 54.94 55.85 58.47 58.69 63.55 65.39 69.72 72.59 74.92 78.96 79.90 83.80 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 5 Rb Sr Y Zr NbMo Tc Ru Rh PdAgCd In Sn Sb Te I Xe 85.47 87.62 88.91 91.22 92.91 95.94 (98) 101.1 102.9 106.4 107.9 112.4 114.8 118.7 121.8 127.6 126.9 131.3 55 56 57 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 6 Cs Ba La* Hf Ta W Re Os Ir Pt AuHg Tl Pb Bi Po At Rn 132.9 137.3 138.9 178.5 180.9 183.9 186.2 190.2 190.2 195.1 197.0 200.5 204.4 207.2 209.0 (210) (210) (222) 87 88 89 104 105 106 107 108 109 110 111 112 114 116 118 7 Fr Ra Ac~RfDb Sg Bh Hs Mt --- --- --- --- --- --- (223) (226) (227) (257) (260) (263) (262) (265) (266) () () () () () () http://pearl1.lanl.gov/periodic/ (1 of 3) [5/17/2001 4:06:20 PM] A Periodic Table of the Elements at Los Alamos National Laboratory 58 59 60 61 62 63 64 65 66 67 68 69 70 71 Lanthanide Series* Ce Pr NdPmSm Eu Gd TbDyHo Er TmYbLu 140.1 140.9 144.2 (147) 150.4 152.0 157.3 158.9 162.5 164.9 167.3 168.9 173.0 175.0 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Actinide Series~ Th Pa U Np Pu AmCmBk Cf Es FmMdNo Lr 232.0 (231) (238) (237) (242) (243) (247) (247) (249) (254) (253) (256) (254) (257) ** Groups are noted by 3 notation conventions. -

Ambient Water Quality Criteria for Silver
United States Office of Water EPA 440/5-80-071 Environmental Protection Regulations and Standards October 1980 Agency Criteria and Standards Division Washington DC 20460 SEPA Ambient Water Quality Criteria for Silver .... AMBIENT WATER QUALITY CRITERIA FOR SILVER Prepared By U.S. ENVIRONMENTAL PROTECTION AGENCY Office of Water Regulations and Standards Criteria and Standards Division Washington, D.C. Office of Research and Development Environmental Criteria and Assessment Office Cincinnati, Ohio Carcinogen Assessment Group Washington, D.C. Environmental Research Laboratories Corvalis, Oregon Duluth, Minnesota Gulf Breeze, Florida Narragansett, Rhode Island , ., " , i DISCLAIMER This report has been reviewed by the Environmental Criteria and Assessment Office, U.S. Environmental Protection Agency, and approved for publication. Mention of trade names or commercial products does not constitute endorsement or recommendation for use. AVAILABILITY NOTICE This document is available to the public through the National Technical Information Service, (NTIS), Springfield, Virginia 22161. :1"':""'"),-;",1 'J " ~ ~.~ .:\, :,," :.l< "'II:: .' " ii FOREWORD Section 304 (a)(1) of the Clean Water Act of 1977 (P.L. 95-217), requires the J\dministrator of the Environmental Protection Agency to publish criter-ia for water quality accurately reflecting the latest scientific kno'w1edge on the kind and extent of all identifiable effects on health and welfare which may be expected from the presence of pollutants in amy body of water, including ground water. Proposed water quality criter"ia for the 65 toxic pollutants listed under section 307 (a)(I) of the Clean Water Act were developed and a notice of their availability wa,s published for public cormnent on March 15, 1979 (44 FR 15926), July 25, 1979 (44 FR 43660), and October 1, 1979 (44 FR 56628).