Identifying the Misshapen Head: Craniosynostosis and Related Disorders Mark S
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PLAGIOCEPHALY and CRANIOSYNOSTOSIS TREATMENT Policy Number: ORT010 Effective Date: February 1, 2019
UnitedHealthcare® Commercial Medical Policy PLAGIOCEPHALY AND CRANIOSYNOSTOSIS TREATMENT Policy Number: ORT010 Effective Date: February 1, 2019 Table of Contents Page COVERAGE RATIONALE ............................................. 1 CENTERS FOR MEDICARE AND MEDICAID SERVICES DEFINITIONS .......................................................... 1 (CMS) .................................................................... 6 APPLICABLE CODES ................................................. 2 REFERENCES .......................................................... 6 DESCRIPTION OF SERVICES ...................................... 2 POLICY HISTORY/REVISION INFORMATION ................. 7 CLINICAL EVIDENCE ................................................ 4 INSTRUCTIONS FOR USE .......................................... 7 U.S. FOOD AND DRUG ADMINISTRATION (FDA) ........... 6 COVERAGE RATIONALE The following are proven and medically necessary: Cranial orthotic devices for treating infants with the following conditions: o Craniofacial asymmetry with severe (non-synostotic) positional plagiocephaly when ALL of the following criteria are met: . Infant is between 3-18 months of age . Severe plagiocephaly is present with or without torticollis . Documentation of a trial of conservative therapy of at least 2 months duration with cranial repositioning, with or without stretching therapy o Craniosynostosis (i.e., synostotic plagiocephaly) following surgical correction Cranial orthotic devices used for treating infants with mild to moderate plagiocephaly -

Coronal and Lambdoid Suture Evolution Following Total Vault Remodeling for Scaphocephaly
NEUROSURGICAL FOCUS Neurosurg Focus 50 (4):E4, 2021 Coronal and lambdoid suture evolution following total vault remodeling for scaphocephaly Pierre-Aurélien Beuriat, MD, PhD,1,2,4 Alexandru Szathmari, MD, PhD,1,2 Julie Chauvel-Picard, MD,1,3,4 Arnaud Gleizal, MD, PhD,1,3,4 Christian Paulus, MD,1,3 Carmine Mottolese, MD, PhD,1,2 and Federico Di Rocco, MD, PhD1,2,4 1French Referral Center for Craniosynostosis; Departments of 2Pediatric Neurosurgery and 3Pediatric Maxillo-Facial Surgery, Hôpital Femme Mère Enfant; and 4Université de Lyon, France OBJECTIVE Different types of surgical procedures are utilized to treat craniosynostosis. In most procedures, the fused suture is removed. There are only a few reports on the evolution of sutures after surgical correction of craniosynostosis. To date, no published study describes neosuture formation after total cranial vault remodeling. The objective of this study was to understand the evolution of the cranial bones in the area of coronal and lambdoid sutures that were removed for complete vault remodeling in patients with sagittal craniosynostosis. In particular, the investigation aimed to confirm the possibility of neosuture formation. METHODS CT images of the skulls of children who underwent operations for scaphocephaly at the Hôpital Femme Mère Enfant, Lyon University Hospital, Lyon, France, from 2004 to 2014 were retrospectively reviewed. Inclusion crite- ria were diagnosis of isolated sagittal synostosis, age between 4 and 18 months at surgery, and availability of reliable postoperative CT images obtained at a minimum of 1 year after surgical correction. Twenty-six boys and 11 girls were included, with a mean age at surgery of 231.6 days (range 126–449 days). -

Frontosphenoidal Synostosis: a Rare Cause of Unilateral Anterior Plagiocephaly
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by RERO DOC Digital Library Childs Nerv Syst (2007) 23:1431–1438 DOI 10.1007/s00381-007-0469-4 ORIGINAL PAPER Frontosphenoidal synostosis: a rare cause of unilateral anterior plagiocephaly Sandrine de Ribaupierre & Alain Czorny & Brigitte Pittet & Bertrand Jacques & Benedict Rilliet Received: 30 March 2007 /Published online: 22 September 2007 # Springer-Verlag 2007 Abstract Conclusion Frontosphenoidal synostosis must be searched Introduction When a child walks in the clinic with a in the absence of a coronal synostosis in a child with unilateral frontal flattening, it is usually associated in our anterior unilateral plagiocephaly, and treated surgically. minds with unilateral coronal synostosis. While the latter might be the most common cause of anterior plagiocephaly, Keywords Craniosynostosis . Pediatric neurosurgery. it is not the only one. A patent coronal suture will force us Anterior plagiocephaly to consider other etiologies, such as deformational plagio- cephaly, or synostosis of another suture. To understand the mechanisms underlying this malformation, the development Introduction and growth of the skull base must be considered. Materials and methods There have been few reports in the Harmonious cranial growth is dependent on patent sutures, literature of isolated frontosphenoidal suture fusion, and and any craniosynostosis might lead to an asymmetrical we would like to report a series of five cases, as the shape of the skull. The anterior skull base is formed of recognition of this entity is important for its treatment. different bones, connected by sutures, fusing at different ages. The frontosphenoidal suture extends from the end of Presented at the Consensus Conference on Pediatric Neurosurgery, the frontoparietal suture, anteriorly and inferiorly in the Rome, 1–2 December 2006. -

Cleidocranial Dysplasia- a Case Report
Case Report Cleidocranial Dysplasia- A Case Report Akhilanand Chaurasia Department of Oral Medicine & Radiology, Faculty of Dental Sciences, King George Medical University, Lucknow E-mail: [email protected] ABSTRACT Cleidocranial dysplasia is a rare congenital disease. It is characterized by autosomal dominant inheritance pattern which is caused due to mutations in the Cbfa1 gene (Runx2) located on chromosome 6p21. It primarily affects bones which are formedby intra-membranous ossification and have equal sex distribution. It is also known as Marie and Sainton disease, Mutational dysostosis and cleidocranialdysostosis. The skeletal deformities of cleidocranial dysplasia are characterized by partial or complete absence of clavicles, late closure of the fontanels, presence of open skull sutures and multiple wormian bones. This rare syndrome is of utmost importance in dentistry due to presence of multiple supernumerary teeth, facial bones deformities and deranged eruption patterns. We are reporting a classical case of cleidocranial dysplasia in 20 year old patient. Keywords: Cleidocranial dysplasia, Marie and Sainton disease, Mutational dysostosis, Cleidocranialdysostosis, Autosomal dominant Access this article online mandibular symphysis are less common findings of Quick Response cleidocranial dysostosis1,10,11. Code: Website: Dental findings in cleidocranialdysostosis are www.innovativepublication.com characterized by a decreased eruptive force of both primary and permanent dentition, prolonged retention of primary teeth12 and an increase in odontogenesis DOI: 10.5958/2395-6194.2015.00009.0 leading to an excessive number of supernumerary teeth13. The clinical findings of cleidocranialdysostosis although present at birth are often either missed or INTRODUCTION diagnosed at a much later time. Cleidocranialdysostosis Cleidocranialdysostosis is a rare congenital defect may be identified by family history, excessive mobility primarily affecting bones which undergo intra- of shoulders and radiographic pathognomonic findings membranous ossification i.e. -

Comparison of the Newborn Skull to the Adult Human Skull
Comparison of the Newborn skull to the Adult Human Skull As a baby grows older their skull goes through a huge change. The neurocranium starts off not as hard as it will be, gaining the ability to shape in whatever way is needed. While their facial cranium, their face, begins to take on unique qualities and changes to look like a mature adult skull. This process takes time but all the changes are very visible. The neurocranium compared to an adult’s is more oval and is substantially bigger than the facial cranium. The newborn's skull has four “horns” two in the front on the frontal bone and two in the back on the parietal bone. These bumps are the thickness that the skull will eventually become. The edges are ridged in between the frontal and the parietal. On top of the skull is the anterior fontanel, which is an opening in the skull that is small and shaped like a diamond. This will close when the child is around two years old. Coming out of the points from the anterior fontanelle are lines or spaces in between the bones, some of these overlap. The advantage of both the spaces in between the bones and the anterior fontanel is room for growth and compression through the birth canal. As a newborn, their neurocranium is 60% of the circumference of an adult’s. At two to three it is 90% of an adult’s, so most of the growth of the neurocranium happens before the child is three. The adult’s skull is more circular and the nose, eyes, and mouth are father apart. -

Morphological and Topographical Study of Wormian Bones in Cadaver Dry Skulls
Original article Morphological and topographical study of Wormian bones in cadaver dry skulls Murlimanju, BV.*, Prabhu, LV., Ashraf, CM., Kumar, CG., Rai, R. and Maheshwari, C. Department of Anatomy, Manipal University, Centre for Basic Sciences, Kasturba Medical College, Mangalore, India *E-mail: [email protected] Abstract Introduction: The Wormian bones are formations associated with insufficient rate of suture closure and regarded as epigenetic and hypostotic traits. It was reported that there exists racial variability among the incidence of these bones. In the present study, the aims were to find the incidence of Wormian bones in Indian skulls and to analyze them topographically. Material and methods: The study included 78 human adult dry skulls of Indian population which were obtained from the neuroanatomy laboratory of our institution. They were macroscopically observed for the incidence and topographical distribution of the Wormian bones. Results: The Wormian bones were observed in 57 skulls (73.1%) of our series. Remaining 21 skulls (26.9%) didn’t show these variant bones. They were observed at the lambdoid suture in 56.4% cases (44 skulls; 14-bilateral; 18-right side; 12-left side), at the asterion in 17.9% (14 skulls; 3-bilateral; 2-right side; 9-left side), at the pterion in 11.5% (9 skulls; 4-right side; 5-left side), at the coronal suture in 1.3% (only one skull) and at the sagittal suture in 1.3% cases (only one skull). Conclusion: The current study observed Wormian bones in 73.1% of the cases from Indian population. This incidence rate is slightly higher compared to other reports and may be due to racial variations. -

Craniosynostosis and Related Disorders Mark S
CLINICAL REPORT Guidance for the Clinician in Rendering Pediatric Care Identifying the Misshapen Head: Craniosynostosis and Related Disorders Mark S. Dias, MD, FAAP, FAANS,a Thomas Samson, MD, FAAP,b Elias B. Rizk, MD, FAAP, FAANS,a Lance S. Governale, MD, FAAP, FAANS,c Joan T. Richtsmeier, PhD,d SECTION ON NEUROLOGIC SURGERY, SECTION ON PLASTIC AND RECONSTRUCTIVE SURGERY Pediatric care providers, pediatricians, pediatric subspecialty physicians, and abstract other health care providers should be able to recognize children with abnormal head shapes that occur as a result of both synostotic and aSection of Pediatric Neurosurgery, Department of Neurosurgery and deformational processes. The purpose of this clinical report is to review the bDivision of Plastic Surgery, Department of Surgery, College of characteristic head shape changes, as well as secondary craniofacial Medicine and dDepartment of Anthropology, College of the Liberal Arts characteristics, that occur in the setting of the various primary and Huck Institutes of the Life Sciences, Pennsylvania State University, State College, Pennsylvania; and cLillian S. Wells Department of craniosynostoses and deformations. As an introduction, the physiology and Neurosurgery, College of Medicine, University of Florida, Gainesville, genetics of skull growth as well as the pathophysiology underlying Florida craniosynostosis are reviewed. This is followed by a description of each type of Clinical reports from the American Academy of Pediatrics benefit from primary craniosynostosis (metopic, unicoronal, bicoronal, sagittal, lambdoid, expertise and resources of liaisons and internal (AAP) and external reviewers. However, clinical reports from the American Academy of and frontosphenoidal) and their resultant head shape changes, with an Pediatrics may not reflect the views of the liaisons or the emphasis on differentiating conditions that require surgical correction from organizations or government agencies that they represent. -
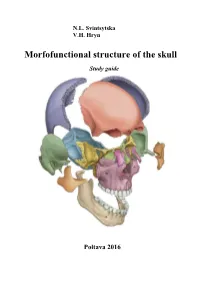
Morfofunctional Structure of the Skull
N.L. Svintsytska V.H. Hryn Morfofunctional structure of the skull Study guide Poltava 2016 Ministry of Public Health of Ukraine Public Institution «Central Methodological Office for Higher Medical Education of MPH of Ukraine» Higher State Educational Establishment of Ukraine «Ukranian Medical Stomatological Academy» N.L. Svintsytska, V.H. Hryn Morfofunctional structure of the skull Study guide Poltava 2016 2 LBC 28.706 UDC 611.714/716 S 24 «Recommended by the Ministry of Health of Ukraine as textbook for English- speaking students of higher educational institutions of the MPH of Ukraine» (minutes of the meeting of the Commission for the organization of training and methodical literature for the persons enrolled in higher medical (pharmaceutical) educational establishments of postgraduate education MPH of Ukraine, from 02.06.2016 №2). Letter of the MPH of Ukraine of 11.07.2016 № 08.01-30/17321 Composed by: N.L. Svintsytska, Associate Professor at the Department of Human Anatomy of Higher State Educational Establishment of Ukraine «Ukrainian Medical Stomatological Academy», PhD in Medicine, Associate Professor V.H. Hryn, Associate Professor at the Department of Human Anatomy of Higher State Educational Establishment of Ukraine «Ukrainian Medical Stomatological Academy», PhD in Medicine, Associate Professor This textbook is intended for undergraduate, postgraduate students and continuing education of health care professionals in a variety of clinical disciplines (medicine, pediatrics, dentistry) as it includes the basic concepts of human anatomy of the skull in adults and newborns. Rewiewed by: O.M. Slobodian, Head of the Department of Anatomy, Topographic Anatomy and Operative Surgery of Higher State Educational Establishment of Ukraine «Bukovinian State Medical University», Doctor of Medical Sciences, Professor M.V. -

Surgical Anatamic of Paranasal Sinuses
SURGICAL ANATAMIC OF PARANASAL SINUSES DR. SEEMA MONGA ASSOCIATE PROFESSOR DEPARTMENT OF ENT-HNS HIMSR MIDDLE TURBINATE 1. Anterior attachment : vertically oriented, sup to the lateral border of cribriform plate. 2. Second attachment :Obliquely oriented- basal lamella/ ground lamella, Attached to the lamina papyracea ( medial wall of orbit anterior, posterior air cells, sphenopala‐ tine foramen 3. Posterior attachment :medial wall of maxillary sinus, horizontally oriented. , supreme turbinate 3. Occasionally 4. fourth turbinate, 5. supreme meatus, if present 6. drains posterior ethmoid drains inferior, middle, superior turbinates and, occasionally, the supreme turbinate, the fourth turbinate. e. Lateral to these turbinates are the corresponding meatuses divided per their drainage systems ANATOMICAL VARIATIONS OF THE TURBINATES 1. Concha bullosa, 24–55%, often bilateral, 2. Interlamellar cell of grunwald: pneumatization is limited to the vertical part of middle turbinate, usually not causing narrowing of the ostiomeatal unit 3. Paradoxic middle turbinate: 26%,. Occasionally, it can affect the patency of the ostiomeatal unit 4. Pneumatized basal lamella, falsely considered, posterior ethmoid air cell Missed basal lamella – attaches to lateral maxillary sinus wall Ostiomeatal unit Anterior ostiomeatal unit, maxillary, anterior ethmoid, frontal sinuses, (1) ethmoid infundibulum, (2) middle meatus, (3) hiatus semilunaris, (4) maxillaryOstium, (5) ethmoid bulla, (6) frontal recess, (7) uncinate process. , sphenoethmoidal recess Other draining osteomeatal unit, posterior in the nasal cavity, posterior ethmoid sinus, lateral to the superior turbinate, . sphenoid Sinus medial to the superior turbinate Uncinate Process Crescent‐shaped, thin individual bone inferiorly- ethmoidal process of inferior turbinate, anterior, lacrimal bone, posteriorly- hiatus Semilunaris, medial -ethmoid infundibulum, laterally, middle meatus superior attachment- variability, direct effect on frontal sinus drainage pathway. -

Blueprint Genetics Craniosynostosis Panel
Craniosynostosis Panel Test code: MA2901 Is a 38 gene panel that includes assessment of non-coding variants. Is ideal for patients with craniosynostosis. About Craniosynostosis Craniosynostosis is defined as the premature fusion of one or more cranial sutures leading to secondary distortion of skull shape. It may result from a primary defect of ossification (primary craniosynostosis) or, more commonly, from a failure of brain growth (secondary craniosynostosis). Premature closure of the sutures (fibrous joints) causes the pressure inside of the head to increase and the skull or facial bones to change from a normal, symmetrical appearance resulting in skull deformities with a variable presentation. Craniosynostosis may occur in an isolated setting or as part of a syndrome with a variety of inheritance patterns and reccurrence risks. Craniosynostosis occurs in 1/2,200 live births. Availability 4 weeks Gene Set Description Genes in the Craniosynostosis Panel and their clinical significance Gene Associated phenotypes Inheritance ClinVar HGMD ALPL Odontohypophosphatasia, Hypophosphatasia perinatal lethal, AD/AR 78 291 infantile, juvenile and adult forms ALX3 Frontonasal dysplasia type 1 AR 8 8 ALX4 Frontonasal dysplasia type 2, Parietal foramina AD/AR 15 24 BMP4 Microphthalmia, syndromic, Orofacial cleft AD 8 39 CDC45 Meier-Gorlin syndrome 7 AR 10 19 EDNRB Hirschsprung disease, ABCD syndrome, Waardenburg syndrome AD/AR 12 66 EFNB1 Craniofrontonasal dysplasia XL 28 116 ERF Craniosynostosis 4 AD 17 16 ESCO2 SC phocomelia syndrome, Roberts syndrome -

CLOSURE of CRANIAL ARTICULATIONS in the SKULI1 of the AUSTRALIAN ABORIGINE by A
CLOSURE OF CRANIAL ARTICULATIONS IN THE SKULI1 OF THE AUSTRALIAN ABORIGINE By A. A. ABBIE, Department of Anatomy, University of Adelaide INTRODUCTION While it is well known that joint closure advances more or less progressively with age, there is still little certainty in matters of detail, mainly for lack of adequate series of documented skulls. In consequence, sundry beliefs have arisen which tend to confuse the issue. One view, now disposed of (see Martin, 1928), is that early suture closure indicates a lower or more primitive type of brain. A corollary, due to Broca (see Topinard, 1890), that the more the brain is exercised the more is suture closure postponed, is equally untenable. A very widespread belief is based on Gratiolet's statement (see Topinard, 1890; Frederic, 1906; Martin, 1928; Fenner, 1939; and others) that in 'lower' skulls the sutures are simple and commence to fuse from in front, while in 'higher' skulls the sutures are more complicated and tend to fuse from behind. This view was disproved by Ribbe (quoted from Frederic, 1906), who substituted the generalization that in dolicocephals synostosis begins in the coronal suture, and in brachycephals in the lambdoid suture. In addition to its purely anthropological interest the subject raises important biological considerations of brain-skull relationship, different foetalization in different ethnological groups (see Bolk, 1926; Weidenreich, 1941; Abbie, 1947), and so on. A survey of the literature reveals very little in the way of data on the age incidence of suture closure. The only substantial contribution accessible here comes from Todd & Lyon (1924) for Europeans, but their work is marred by arbitrary rejection of awkward material. -

MECHANISMS in ENDOCRINOLOGY: Novel Genetic Causes of Short Stature
J M Wit and others Genetics of short stature 174:4 R145–R173 Review MECHANISMS IN ENDOCRINOLOGY Novel genetic causes of short stature 1 1 2 2 Jan M Wit , Wilma Oostdijk , Monique Losekoot , Hermine A van Duyvenvoorde , Correspondence Claudia A L Ruivenkamp2 and Sarina G Kant2 should be addressed to J M Wit Departments of 1Paediatrics and 2Clinical Genetics, Leiden University Medical Center, PO Box 9600, 2300 RC Leiden, Email The Netherlands [email protected] Abstract The fast technological development, particularly single nucleotide polymorphism array, array-comparative genomic hybridization, and whole exome sequencing, has led to the discovery of many novel genetic causes of growth failure. In this review we discuss a selection of these, according to a diagnostic classification centred on the epiphyseal growth plate. We successively discuss disorders in hormone signalling, paracrine factors, matrix molecules, intracellular pathways, and fundamental cellular processes, followed by chromosomal aberrations including copy number variants (CNVs) and imprinting disorders associated with short stature. Many novel causes of GH deficiency (GHD) as part of combined pituitary hormone deficiency have been uncovered. The most frequent genetic causes of isolated GHD are GH1 and GHRHR defects, but several novel causes have recently been found, such as GHSR, RNPC3, and IFT172 mutations. Besides well-defined causes of GH insensitivity (GHR, STAT5B, IGFALS, IGF1 defects), disorders of NFkB signalling, STAT3 and IGF2 have recently been discovered. Heterozygous IGF1R defects are a relatively frequent cause of prenatal and postnatal growth retardation. TRHA mutations cause a syndromic form of short stature with elevated T3/T4 ratio. Disorders of signalling of various paracrine factors (FGFs, BMPs, WNTs, PTHrP/IHH, and CNP/NPR2) or genetic defects affecting cartilage extracellular matrix usually cause disproportionate short stature.