WO 2009/133538 Al
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Review of Market for Octane Enhancers
May 2000 • NREL/SR-580-28193 Review of Market for Octane Enhancers Final Report J.E. Sinor Consultants, Inc. Niwot, Colorado National Renewable Energy Laboratory 1617 Cole Boulevard Golden, Colorado 80401-3393 NREL is a U.S. Department of Energy Laboratory Operated by Midwest Research Institute • Battelle • Bechtel Contract No. DE-AC36-99-GO10337 May 2000 • NREL/SR-580-28193 Review of Market for Octane Enhancers Final Report J.E. Sinor Consultants, Inc. Niwot, Colorado NREL Technical Monitor: K. Ibsen Prepared under Subcontract No. TXE-0-29113-01 National Renewable Energy Laboratory 1617 Cole Boulevard Golden, Colorado 80401-3393 NREL is a U.S. Department of Energy Laboratory Operated by Midwest Research Institute • Battelle • Bechtel Contract No. DE-AC36-99-GO10337 NOTICE This report was prepared as an account of work sponsored by an agency of the United States government. Neither the United States government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States government or any agency thereof. Available electronically at http://www.doe.gov/bridge Available for a processing fee to U.S. -

Precursors and Chemicals Frequently Used in the Illicit Manufacture of Narcotic Drugs and Psychotropic Substances 2017
INTERNATIONAL NARCOTICS CONTROL BOARD Precursors and chemicals frequently used in the illicit manufacture of narcotic drugs and psychotropic substances 2017 EMBARGO Observe release date: Not to be published or broadcast before Thursday, 1 March 2018, at 1100 hours (CET) UNITED NATIONS CAUTION Reports published by the International Narcotics Control Board in 2017 The Report of the International Narcotics Control Board for 2017 (E/INCB/2017/1) is supplemented by the following reports: Narcotic Drugs: Estimated World Requirements for 2018—Statistics for 2016 (E/INCB/2017/2) Psychotropic Substances: Statistics for 2016—Assessments of Annual Medical and Scientific Requirements for Substances in Schedules II, III and IV of the Convention on Psychotropic Substances of 1971 (E/INCB/2017/3) Precursors and Chemicals Frequently Used in the Illicit Manufacture of Narcotic Drugs and Psychotropic Substances: Report of the International Narcotics Control Board for 2017 on the Implementation of Article 12 of the United Nations Convention against Illicit Traffic in Narcotic Drugs and Psychotropic Substances of 1988 (E/INCB/2017/4) The updated lists of substances under international control, comprising narcotic drugs, psychotropic substances and substances frequently used in the illicit manufacture of narcotic drugs and psychotropic substances, are contained in the latest editions of the annexes to the statistical forms (“Yellow List”, “Green List” and “Red List”), which are also issued by the Board. Contacting the International Narcotics Control Board The secretariat of the Board may be reached at the following address: Vienna International Centre Room E-1339 P.O. Box 500 1400 Vienna Austria In addition, the following may be used to contact the secretariat: Telephone: (+43-1) 26060 Fax: (+43-1) 26060-5867 or 26060-5868 Email: [email protected] The text of the present report is also available on the website of the Board (www.incb.org). -

Organocatalytic Asymmetric N-Sulfonyl Amide C-N Bond Activation to Access Axially Chiral Biaryl Amino Acids
ARTICLE https://doi.org/10.1038/s41467-020-14799-8 OPEN Organocatalytic asymmetric N-sulfonyl amide C-N bond activation to access axially chiral biaryl amino acids Guanjie Wang1, Qianqian Shi2, Wanyao Hu1, Tao Chen1, Yingying Guo1, Zhouli Hu1, Minghua Gong1, ✉ ✉ ✉ Jingcheng Guo1, Donghui Wei 2 , Zhenqian Fu 1,3 & Wei Huang1,3 1234567890():,; Amides are among the most fundamental functional groups and essential structural units, widely used in chemistry, biochemistry and material science. Amide synthesis and trans- formations is a topic of continuous interest in organic chemistry. However, direct catalytic asymmetric activation of amide C-N bonds still remains a long-standing challenge due to high stability of amide linkages. Herein, we describe an organocatalytic asymmetric amide C-N bonds cleavage of N-sulfonyl biaryl lactams under mild conditions, developing a general and practical method for atroposelective construction of axially chiral biaryl amino acids. A structurally diverse set of axially chiral biaryl amino acids are obtained in high yields with excellent enantioselectivities. Moreover, a variety of axially chiral unsymmetrical biaryl organocatalysts are efficiently constructed from the resulting axially chiral biaryl amino acids by our present strategy, and show competitive outcomes in asymmetric reactions. 1 Key Laboratory of Flexible Electronics & Institute of Advanced Materials, Jiangsu National Synergetic Innovation Center for Advanced Materials, Nanjing Tech University, 30 South Puzhu Road, Nanjing 211816, China. 2 College -
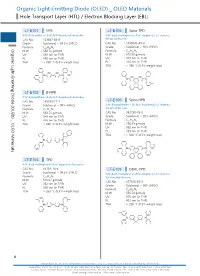
Organic Light-Emitting Diode (OLED) OLED Materials Hole Transport Layer (HTL) / Electron Blocking Layer (EBL)
Organic Light-EmittingOrganic Light-Emitting DiodeHole Transport(OLED) Layer (HTL) / Diode (OLED) _ OLED Materials Electron Blocking Layer (EBL) Organic Light-Emitting Diode (OLED) _ OLED Materials Hole Transport Layer (HTL) / Electron Blocking Layer (EBL) LT-E101 NPB LT-E105 Spiro-TPD N,N'-Bis(naphthalen-1-yl)-N,N'-bis(phenyl)-benzidine N,N'-Bis(3-methylphenyl)-N,N'-bis(phenyl)-2,7-diamino- CAS No. : 123847-85-8 9,9-spirobifluorene Grade : Sublimed, > 99.5% (HPLC) CAS No. : 1033035-83-4 : Grade : Sublimed, > 99% (HPLC) Organic Light-Emitting Diode (OLED) _ OLED Materials Formula C44H32N2 M.W. : 588.74 g/mole Formula : C51H38N2 UV : 339 nm (in THF) M.W. : 678.86 g/mole PL : 450 nm (in THF) UV : 379 nm (in THF) TGA : > 350 °C (0.5% weight loss) PL : 416 nm (in THF) TGA : > 280 °C (0.5% weight loss) N N N N LT-E102 β-NPB N,N'-Bis(naphthalen-2-yl)-N,N'-bis(phenyl)-benzidine Spiro-NPB CAS No. : 139255-17-7 LT-E106 Grade : Sublimed, > 99% (HPLC) N,N'-Bis(naphthalen-1-yl)-N,N'-bis(phenyl)-2,7-diamino- 9,9-spirobifluorene Formula : C44H32N2 M.W. : 588.74 g/mole CAS No. : 932739-76-9 UV : 349 nm (in THF) Grade : Sublimed, > 99% (HPLC) PL : 416 nm (in THF) Formula : C57H38N2 TGA : > 330 °C (0.5% weight loss) M.W. : 750.93 g/mole UV : 380 nm (in THF) PL : 453 nm (in THF) TGA : > 360 °C (0.5% weight loss) N N N N LT-E103 TPD N,N'-Bis(3-methylphenyl)-N,N'-bis(phenyl)-benzidine CAS No. -

Hydroxydopamine Lesions of the Nucleus Accumbens and the Mglur2/3 Agonist LY379268
Neuropsychopharmacology (2003) 28, 1440–1447 & 2003 Nature Publishing Group All rights reserved 0893-133X/03 $25.00 www.neuropsychopharmacology.org Toluene-Induced Locomotor Activity is Blocked by 6- Hydroxydopamine Lesions of the Nucleus Accumbens and the mGluR2/3 Agonist LY379268 1,3 2 ,1 AC Riegel , SF Ali , ED French* 1Department of Pharmacology, College of Medicine, University of Arizona, Tucson, AZ, USA; 2Neurochemistry Laboratory, Division of Neurotoxicology, National Center for Toxicological Research/US FDA, Jefferson, AR, USA The abuse of volatile inhalants remains a prominent, yet poorly understood, form of substance abuse among youth. Nevertheless, the identification of a mechanism underlying the reinforcing properties of inhalants has been hampered by the lack of a clearly identifiable neural substrate upon which these chemicals act. One ingredient that is common to many abused inhalants is toluene, an organic solvent that is self-administered by nonhuman primates and rodents. Most drugs of abuse have been found to elicit forward locomotion in rats, an effect owing to the activation of mesoaccumbal dopamine (DA) pathways. Thus, the present study was undertaken using two different approaches to determine whether toluene-induced locomotor hyperactivity is also ultimately dependent upon DA neurotransmission in the mesolimbic nucleus accumbens (NAC). Here we report on the effects of 6-hydroxydopamine (6-OHDA) lesions of the NAC or pretreatment with the metabotropic mGlu2/3 receptor agonist LY379268 on toluene-induced locomotor activity. Both procedures, which are known to alter neurotransmission within the NAC, significantly attenuated toluene’s locomotor stimulatory effects. These results provide strong support for a central mechanism of action of inhalants, which in the past has been more typically attributed to general nonspecific mechanisms throughout the brain. -

Inhalant Abuse Pediatric Care
CLINICAL REPORT Guidance for the Clinician in Rendering Inhalant Abuse Pediatric Care Janet F. Williams, MD, Michael Storck, MD, and the Committee on Substance Abuse and Committee on Native American Child Health ABSTRACT Inhalant abuse is the intentional inhalation of a volatile substance for the purpose of achieving an altered mental state. As an important, yet-underrecognized form of substance abuse, inhalant abuse crosses all demographic, ethnic, and socioeco- nomic boundaries, causing significant morbidity and mortality in school-aged and older children. This clinical report reviews key aspects of inhalant abuse, empha- sizes the need for greater awareness, and offers advice regarding the pediatrician’s role in the prevention and management of this substance abuse problem. TYPES OF CHEMICALS AND PRODUCTS ABUSED The term “inhalant” encompasses a wide range of pharmacologically diverse substances that readily vaporize. Most other substances of abuse are classified by grouping together substances that share a specific central nervous system action or perceived psychoactive effect, but inhalant substances that are abused are grouped by having a common route of drug use. Inhalant abuse, sometimes referred to as solvent or volatile substance abuse, can be better understood when the expansive list of inhalants is classified into 3 groups on the basis of what is currently known pharmacologically: group I includes volatile solvents, fuels, and anesthetics; group II includes nitrous oxide; and group III includes volatile alkyl nitrites (Table 1). This classification is also consistent with reported differences in user populations, patterns of abuse, and associated problems seen clinically.1–3 Drugs that do not www.pediatrics.org/cgi/doi/10.1542/ readily vaporize at room temperature, such as cocaine, heroin, nicotine, or alcohol, peds.2007-0470 can also be abused through inhalation, but characteristic pharmacologic properties doi:10.1542/peds.2007-0470 distinguish these substances from inhalants. -

The Effects of the Morphine Analogue Levorphanol on Leukocytes: Metabolic Effects at Rest and During Phagocytosis
The effects of the morphine analogue levorphanol on leukocytes: Metabolic effects at rest and during phagocytosis Nancy Wurster, … , Penelope Pettis, Sharon Lebow J Clin Invest. 1971;50(5):1091-1099. https://doi.org/10.1172/JCI106580. Studies on bacteria have suggested that morphine-like drugs have effects on the cell membrane. To determine the effect of this class of drugs on a mammalian cell, we selected the rabbit peritoneal exudate granulocyte, which undergoes striking membrane changes during phagocytosis. We examined the effect in vitro of the morphine analogue, levorphanol on phagocytosis and metabolism by granulocytes incubated with and without polystyrene particles or live Escherichia coli. Levorphanol (1 or 2 mmoles/liter) decreased: (a) acylation of lysolecithin or lysophosphatidylethanolamine in the medium (which is stimulated about two-fold during phagocytosis) both at rest (40%) and during phagocytosis (60%); (b) uptake of latex particles and Escherichia coli, as judged by electron microscopy; (c) killing of live Escherichia coli (10-fold); (d) 14 14 + CO2 production from glucose-1- C during phagocytosis by at least 80%; (e) K content of granulocytes (35%); (f) oxidation of linoleate-1-14C by 50%, and its incorporation into triglyceride by more than 80%. However, levorphanol stimulated 2 to 3-fold the incorporation of linoleate-1-14C or palmitate-1-14C into several phospholipids. Glucose uptake, lactate production, and adenosine triphosphate (ATP) content are not affected by the drug. Thus, levorphanol does not appear to exert its effects through generalized metabolic suppression. Removal of levorphanol by twice resuspending the granulocytes completely reverses all inhibition. In line with observations on bacteria, it appears that the complex effects of levorphanol on […] Find the latest version: https://jci.me/106580/pdf The Effects of the Morphine Analogue Levorphanol on Leukocytes METABOLIC EFFECTS AT REST AND DURING PHAGOCYTOSIS NANcY WuRsTE, PETER ELSBACH, ERIc J. -

Consultation Documents
WHO Drug Information Vol. 29, No. 3, 2015 Consultation documents To receive draft monographs by email please contact Mrs Wendy Bonny ([email protected]), specifying that you wish to be added to the electronic mailing list. The International Pharmacopoeia Carbamazepinum Carbamazepine This is a draft proposal for The International Pharmacopoeia (Working document QAS/15.608, July 2015). The working document with line numbers and tracked changes is available for comment at www.who.int/medicines/areas/quality_safety/quality_assurance/projects/en/. Please address any comments to: World Health Organization, Quality Assurance and Safety: Medicines, Dr Herbert Schmidt, 1211 Geneva 27, Switzerland; fax: +41 22 791 4730; email: [email protected]. [Note from the Secretariat. It is proposed to revise the monograph on Carbamazepine in The International Pharmacopoeia.] [Note from the editor. In accordance with WHO editorial policy the text reproduced below does not include tracked changes. Changes from the current monograph are indicated by insert and delete in the working document available at the above-mentioned web address.] Molecular formula. C15H12N2O Relative molecular mass. 236.3 Graphic formula. Chemical name. 5H-Dibenz[b,f]azepine-5-carboxamide; CAS Reg. No. 298-46-4. Description. A white to almost white, crystalline powder. Solubility. Practically insoluble in water; sparingly soluble in acetone; soluble in ethanol (~750 g/L) TS; freely soluble in dichloromethane. Category. Antiepileptic. 352 WHO Drug Information Vol. 29, No. 3, 2015 Consultation documents Additional information. Carbamazepine exhibits polymorphism. The acceptable crystalline form is anhydrous polymorph form III1. It corresponds to carbamazepine RS. Storage. Carbamazepine should be kept in a tightly closed container. -

Direct Analysis of Psilocin and Muscimol in Urine Samples Using Single Drop Microextraction Technique In-Line with Capillary Electrophoresis
molecules Article Direct Analysis of Psilocin and Muscimol in Urine Samples Using Single Drop Microextraction Technique In-Line with Capillary Electrophoresis Anna Poliwoda * , Katarzyna Zieli ´nskaand Piotr P. Wieczorek Faculty of Chemistry, University of Opole, Oleska 48, 45-042 Opole, Poland * Correspondence: [email protected]; Tel.: +48-77452-7116 Academic Editors: Mihkel Koel and Marek Tobiszewski Received: 20 February 2020; Accepted: 25 March 2020; Published: 29 March 2020 Abstract: The fully automated system of single drop microextraction coupled with capillary electrophoresis (SDME-CE) was developed for in-line preconcentration and determination of muscimol (MUS) and psilocin (PSC) from urine samples. Those two analytes are characteristic active metabolites of Amanita and Psilocybe mushrooms, evoking visual and auditory hallucinations. Study analytes were selectively extracted from the donor phase (urine samples, pH 4) into the organic phase (a drop of octanol layer), and re-extracted to the acidic acceptor (background electrolyte, BGE), consisting of 25 mM phosphate buffer (pH 3). The optimized conditions for the extraction procedure of a 200 µL urine sample allowed us to obtain more than a 170-fold enrichment effect. The calibration curves 1 were linear in the range of 0.05–50 mg L− , with the correlation coefficients from 0.9911 to 0.9992. The limit of detections was determined by spiking blank urine samples with appropriate standards, 1 1 i.e., 0.004 mg L− for PSC and 0.016 mg L− for MUS, respectively. The limits of quantification varied 1 1 from 0.014 mg L− for PSC and 0.045 mg L− for MUS. -

Drugs of Abuseon September Archived 13-10048 No
U.S. DEPARTMENT OF JUSTICE DRUG ENFORCEMENT ADMINISTRATION WWW.DEA.GOV 9, 2014 on September archived 13-10048 No. v. Stewart, in U.S. cited Drugs of2011 Abuse EDITION A DEA RESOURCE GUIDE V. Narcotics WHAT ARE NARCOTICS? Also known as “opioids,” the term "narcotic" comes from the Greek word for “stupor” and originally referred to a variety of substances that dulled the senses and relieved pain. Though some people still refer to all drugs as “narcot- ics,” today “narcotic” refers to opium, opium derivatives, and their semi-synthetic substitutes. A more current term for these drugs, with less uncertainty regarding its meaning, is “opioid.” Examples include the illicit drug heroin and pharmaceutical drugs like OxyContin®, Vicodin®, codeine, morphine, methadone and fentanyl. WHAT IS THEIR ORIGIN? The poppy papaver somniferum is the source for all natural opioids, whereas synthetic opioids are made entirely in a lab and include meperidine, fentanyl, and methadone. Semi-synthetic opioids are synthesized from naturally occurring opium products, such as morphine and codeine, and include heroin, oxycodone, hydrocodone, and hydromorphone. Teens can obtain narcotics from friends, family members, medicine cabinets, pharmacies, nursing 2014 homes, hospitals, hospices, doctors, and the Internet. 9, on September archived 13-10048 No. v. Stewart, in U.S. cited What are common street names? Street names for various narcotics/opioids include: ➔ Hillbilly Heroin, Lean or Purple Drank, OC, Ox, Oxy, Oxycotton, Sippin Syrup What are their forms? Narcotics/opioids come in various forms including: ➔ T ablets, capsules, skin patches, powder, chunks in varying colors (from white to shades of brown and black), liquid form for oral use and injection, syrups, suppositories, lollipops How are they abused? ➔ Narcotics/opioids can be swallowed, smoked, sniffed, or injected. -

Medial Prefrontal Cortex Inversely Regulates Toluene-Induced Changes in Markers of Synaptic Plasticity of Mesolimbic Dopamine Neurons
804 • The Journal of Neuroscience, January 9, 2013 • 33(2):804–813 Systems/Circuits Medial Prefrontal Cortex Inversely Regulates Toluene-Induced Changes in Markers of Synaptic Plasticity of Mesolimbic Dopamine Neurons Jacob T. Beckley, Caitlin E. Evins, Hleb Fedarovich, Meghin J. Gilstrap, and John J. Woodward Department of Neurosciences, and Center for Drug and Alcohol Programs, Department of Psychiatry, Medical University of South Carolina, Charleston, South Carolina 29425 Toluene is a volatile solvent that is intentionally inhaled by children, adolescents, and adults for its intoxicating effects. Although voluntary use of toluene suggests that it possesses rewarding properties and abuse potential, it is unknown whether toluene alters excitatory synaptic transmission in reward-sensitive dopamine neurons like other drugs of abuse. Here, using a combination of retro- grade labeling and slice electrophysiology, we show that a brief in vivo exposure of rats to a behaviorally relevant concentration of toluene vapor enhances glutamatergic synaptic strength of dopamine (DA) neurons projecting to nucleus accumbens core and medial shell neurons. This effect persisted for up to3dinmesoaccumbens core DA neurons and for at least 21 d in those projecting to the medial shell. In contrast, toluene vapor exposure had no effect on synaptic strength of DA neurons that project to the medial prefrontal cortex (mPFC). Furthermore, infusion of GABAergic modulators into the mPFC before vapor exposure to pharmacologically manipulate output, inhib- ited, or potentiated toluene’s action on mesoaccumbens DA neurons. Together, the results of these studies indicate that toluene induces a target-selective increase in mesolimbic DA neuron synaptic transmission and strongly implicates the mPFC as an important regulator of drug-induced plasticity of mesolimbic dopamine neurons. -

Chemicals Required for the Illicit Manufacture of Drugs Table 1 SUBSTANCES in TABLES I and II of the 1988 CONVENTION
Chemicals Required 1. A variety of chemicals are used in the illicit manufacture of for the Illicit drugs. The United Nations Convention against Illicit Traffic in Manufacture of Drugs Narcotic Drugs and Psychotropic Substances of 1988 (1988 Convention) refers to “substances frequently used in the illicit manufacture of narcotic drugs and psychotropic substances”. Twenty-two such substances are listed in Tables I and II of the 1988 Convention as in force on 1st May, 1998. (See Table1) Table 1 Table I Table II SUBSTANCES IN N-Acetylanthranilic acid. Acetic anhydride TABLES I AND II OF Ephedrine Acetone THE 1988 Ergometrine Anthranilic acid CONVENTION Ergotamine Ethyl ether Isosafrole Hydrochloric acid* Lysergic acid Methyl ethyl ketone 3,4-methylenedioxyphenyl-2-propanone Phenylacetic acid 1-phenyl-2-propanone Piperidine Piperonal Potassium permanganate Pseudoephedrine Sulphuric acid* Safrole Toluene The salts of the substances in this Table The salts of the substances whenever the existence of such salts is in this Table whenever the possible. existence of such salts is possible. * The salts of hydrochloric acid and sulphuric acid are specifically excluded from Table II. U N D C P 11 The term “precursor” is used to indicate any of these substances in the two Tables. Chemicals used in the illicit manufacture of narcotic drugs and psychotropic substances are often described as precursors or essential chemicals, and these include true precursors, solvents, oxidising agents and other Chemicals used in the illicit manufacture of narcotic substances. Although the term is not drugs and psychotropic substances are often technically correct, it has become common described as precursors or essential chemicals, and practice to refer to all such substances as these include true precursors, solvents, oxidising “precursors”.