Direct Analysis of Psilocin and Muscimol in Urine Samples Using Single Drop Microextraction Technique In-Line with Capillary Electrophoresis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Review of Market for Octane Enhancers
May 2000 • NREL/SR-580-28193 Review of Market for Octane Enhancers Final Report J.E. Sinor Consultants, Inc. Niwot, Colorado National Renewable Energy Laboratory 1617 Cole Boulevard Golden, Colorado 80401-3393 NREL is a U.S. Department of Energy Laboratory Operated by Midwest Research Institute • Battelle • Bechtel Contract No. DE-AC36-99-GO10337 May 2000 • NREL/SR-580-28193 Review of Market for Octane Enhancers Final Report J.E. Sinor Consultants, Inc. Niwot, Colorado NREL Technical Monitor: K. Ibsen Prepared under Subcontract No. TXE-0-29113-01 National Renewable Energy Laboratory 1617 Cole Boulevard Golden, Colorado 80401-3393 NREL is a U.S. Department of Energy Laboratory Operated by Midwest Research Institute • Battelle • Bechtel Contract No. DE-AC36-99-GO10337 NOTICE This report was prepared as an account of work sponsored by an agency of the United States government. Neither the United States government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States government or any agency thereof. Available electronically at http://www.doe.gov/bridge Available for a processing fee to U.S. -

Precursors and Chemicals Frequently Used in the Illicit Manufacture of Narcotic Drugs and Psychotropic Substances 2017
INTERNATIONAL NARCOTICS CONTROL BOARD Precursors and chemicals frequently used in the illicit manufacture of narcotic drugs and psychotropic substances 2017 EMBARGO Observe release date: Not to be published or broadcast before Thursday, 1 March 2018, at 1100 hours (CET) UNITED NATIONS CAUTION Reports published by the International Narcotics Control Board in 2017 The Report of the International Narcotics Control Board for 2017 (E/INCB/2017/1) is supplemented by the following reports: Narcotic Drugs: Estimated World Requirements for 2018—Statistics for 2016 (E/INCB/2017/2) Psychotropic Substances: Statistics for 2016—Assessments of Annual Medical and Scientific Requirements for Substances in Schedules II, III and IV of the Convention on Psychotropic Substances of 1971 (E/INCB/2017/3) Precursors and Chemicals Frequently Used in the Illicit Manufacture of Narcotic Drugs and Psychotropic Substances: Report of the International Narcotics Control Board for 2017 on the Implementation of Article 12 of the United Nations Convention against Illicit Traffic in Narcotic Drugs and Psychotropic Substances of 1988 (E/INCB/2017/4) The updated lists of substances under international control, comprising narcotic drugs, psychotropic substances and substances frequently used in the illicit manufacture of narcotic drugs and psychotropic substances, are contained in the latest editions of the annexes to the statistical forms (“Yellow List”, “Green List” and “Red List”), which are also issued by the Board. Contacting the International Narcotics Control Board The secretariat of the Board may be reached at the following address: Vienna International Centre Room E-1339 P.O. Box 500 1400 Vienna Austria In addition, the following may be used to contact the secretariat: Telephone: (+43-1) 26060 Fax: (+43-1) 26060-5867 or 26060-5868 Email: [email protected] The text of the present report is also available on the website of the Board (www.incb.org). -

Organocatalytic Asymmetric N-Sulfonyl Amide C-N Bond Activation to Access Axially Chiral Biaryl Amino Acids
ARTICLE https://doi.org/10.1038/s41467-020-14799-8 OPEN Organocatalytic asymmetric N-sulfonyl amide C-N bond activation to access axially chiral biaryl amino acids Guanjie Wang1, Qianqian Shi2, Wanyao Hu1, Tao Chen1, Yingying Guo1, Zhouli Hu1, Minghua Gong1, ✉ ✉ ✉ Jingcheng Guo1, Donghui Wei 2 , Zhenqian Fu 1,3 & Wei Huang1,3 1234567890():,; Amides are among the most fundamental functional groups and essential structural units, widely used in chemistry, biochemistry and material science. Amide synthesis and trans- formations is a topic of continuous interest in organic chemistry. However, direct catalytic asymmetric activation of amide C-N bonds still remains a long-standing challenge due to high stability of amide linkages. Herein, we describe an organocatalytic asymmetric amide C-N bonds cleavage of N-sulfonyl biaryl lactams under mild conditions, developing a general and practical method for atroposelective construction of axially chiral biaryl amino acids. A structurally diverse set of axially chiral biaryl amino acids are obtained in high yields with excellent enantioselectivities. Moreover, a variety of axially chiral unsymmetrical biaryl organocatalysts are efficiently constructed from the resulting axially chiral biaryl amino acids by our present strategy, and show competitive outcomes in asymmetric reactions. 1 Key Laboratory of Flexible Electronics & Institute of Advanced Materials, Jiangsu National Synergetic Innovation Center for Advanced Materials, Nanjing Tech University, 30 South Puzhu Road, Nanjing 211816, China. 2 College -
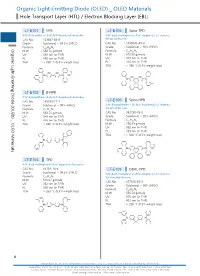
Organic Light-Emitting Diode (OLED) OLED Materials Hole Transport Layer (HTL) / Electron Blocking Layer (EBL)
Organic Light-EmittingOrganic Light-Emitting DiodeHole Transport(OLED) Layer (HTL) / Diode (OLED) _ OLED Materials Electron Blocking Layer (EBL) Organic Light-Emitting Diode (OLED) _ OLED Materials Hole Transport Layer (HTL) / Electron Blocking Layer (EBL) LT-E101 NPB LT-E105 Spiro-TPD N,N'-Bis(naphthalen-1-yl)-N,N'-bis(phenyl)-benzidine N,N'-Bis(3-methylphenyl)-N,N'-bis(phenyl)-2,7-diamino- CAS No. : 123847-85-8 9,9-spirobifluorene Grade : Sublimed, > 99.5% (HPLC) CAS No. : 1033035-83-4 : Grade : Sublimed, > 99% (HPLC) Organic Light-Emitting Diode (OLED) _ OLED Materials Formula C44H32N2 M.W. : 588.74 g/mole Formula : C51H38N2 UV : 339 nm (in THF) M.W. : 678.86 g/mole PL : 450 nm (in THF) UV : 379 nm (in THF) TGA : > 350 °C (0.5% weight loss) PL : 416 nm (in THF) TGA : > 280 °C (0.5% weight loss) N N N N LT-E102 β-NPB N,N'-Bis(naphthalen-2-yl)-N,N'-bis(phenyl)-benzidine Spiro-NPB CAS No. : 139255-17-7 LT-E106 Grade : Sublimed, > 99% (HPLC) N,N'-Bis(naphthalen-1-yl)-N,N'-bis(phenyl)-2,7-diamino- 9,9-spirobifluorene Formula : C44H32N2 M.W. : 588.74 g/mole CAS No. : 932739-76-9 UV : 349 nm (in THF) Grade : Sublimed, > 99% (HPLC) PL : 416 nm (in THF) Formula : C57H38N2 TGA : > 330 °C (0.5% weight loss) M.W. : 750.93 g/mole UV : 380 nm (in THF) PL : 453 nm (in THF) TGA : > 360 °C (0.5% weight loss) N N N N LT-E103 TPD N,N'-Bis(3-methylphenyl)-N,N'-bis(phenyl)-benzidine CAS No. -

Toxicological and Pharmacological Profile of Amanita Muscaria (L.) Lam
Pharmacia 67(4): 317–323 DOI 10.3897/pharmacia.67.e56112 Review Article Toxicological and pharmacological profile of Amanita muscaria (L.) Lam. – a new rising opportunity for biomedicine Maria Voynova1, Aleksandar Shkondrov2, Magdalena Kondeva-Burdina1, Ilina Krasteva2 1 Laboratory of Drug metabolism and drug toxicity, Department “Pharmacology, Pharmacotherapy and Toxicology”, Faculty of Pharmacy, Medical University of Sofia, Bulgaria 2 Department of Pharmacognosy, Faculty of Pharmacy, Medical University of Sofia, Bulgaria Corresponding author: Magdalena Kondeva-Burdina ([email protected]) Received 2 July 2020 ♦ Accepted 19 August 2020 ♦ Published 26 November 2020 Citation: Voynova M, Shkondrov A, Kondeva-Burdina M, Krasteva I (2020) Toxicological and pharmacological profile of Amanita muscaria (L.) Lam. – a new rising opportunity for biomedicine. Pharmacia 67(4): 317–323. https://doi.org/10.3897/pharmacia.67. e56112 Abstract Amanita muscaria, commonly known as fly agaric, is a basidiomycete. Its main psychoactive constituents are ibotenic acid and mus- cimol, both involved in ‘pantherina-muscaria’ poisoning syndrome. The rising pharmacological and toxicological interest based on lots of contradictive opinions concerning the use of Amanita muscaria extracts’ neuroprotective role against some neurodegenerative diseases such as Parkinson’s and Alzheimer’s, its potent role in the treatment of cerebral ischaemia and other socially significant health conditions gave the basis for this review. Facts about Amanita muscaria’s morphology, chemical content, toxicological and pharmacological characteristics and usage from ancient times to present-day’s opportunities in modern medicine are presented. Keywords Amanita muscaria, muscimol, ibotenic acid Introduction rica, the genus had an ancestral origin in the Siberian-Be- ringian region in the Tertiary period (Geml et al. -

Hydroxydopamine Lesions of the Nucleus Accumbens and the Mglur2/3 Agonist LY379268
Neuropsychopharmacology (2003) 28, 1440–1447 & 2003 Nature Publishing Group All rights reserved 0893-133X/03 $25.00 www.neuropsychopharmacology.org Toluene-Induced Locomotor Activity is Blocked by 6- Hydroxydopamine Lesions of the Nucleus Accumbens and the mGluR2/3 Agonist LY379268 1,3 2 ,1 AC Riegel , SF Ali , ED French* 1Department of Pharmacology, College of Medicine, University of Arizona, Tucson, AZ, USA; 2Neurochemistry Laboratory, Division of Neurotoxicology, National Center for Toxicological Research/US FDA, Jefferson, AR, USA The abuse of volatile inhalants remains a prominent, yet poorly understood, form of substance abuse among youth. Nevertheless, the identification of a mechanism underlying the reinforcing properties of inhalants has been hampered by the lack of a clearly identifiable neural substrate upon which these chemicals act. One ingredient that is common to many abused inhalants is toluene, an organic solvent that is self-administered by nonhuman primates and rodents. Most drugs of abuse have been found to elicit forward locomotion in rats, an effect owing to the activation of mesoaccumbal dopamine (DA) pathways. Thus, the present study was undertaken using two different approaches to determine whether toluene-induced locomotor hyperactivity is also ultimately dependent upon DA neurotransmission in the mesolimbic nucleus accumbens (NAC). Here we report on the effects of 6-hydroxydopamine (6-OHDA) lesions of the NAC or pretreatment with the metabotropic mGlu2/3 receptor agonist LY379268 on toluene-induced locomotor activity. Both procedures, which are known to alter neurotransmission within the NAC, significantly attenuated toluene’s locomotor stimulatory effects. These results provide strong support for a central mechanism of action of inhalants, which in the past has been more typically attributed to general nonspecific mechanisms throughout the brain. -

Amanita Muscaria: Ecology, Chemistry, Myths
Entry Amanita muscaria: Ecology, Chemistry, Myths Quentin Carboué * and Michel Lopez URD Agro-Biotechnologies Industrielles (ABI), CEBB, AgroParisTech, 51110 Pomacle, France; [email protected] * Correspondence: [email protected] Definition: Amanita muscaria is the most emblematic mushroom in the popular representation. It is an ectomycorrhizal fungus endemic to the cold ecosystems of the northern hemisphere. The basidiocarp contains isoxazoles compounds that have specific actions on the central nervous system, including hallucinations. For this reason, it is considered an important entheogenic mushroom in different cultures whose remnants are still visible in some modern-day European traditions. In Siberian civilizations, it has been consumed for religious and recreational purposes for millennia, as it was the only inebriant in this region. Keywords: Amanita muscaria; ibotenic acid; muscimol; muscarine; ethnomycology 1. Introduction Thanks to its peculiar red cap with white spots, Amanita muscaria (L.) Lam. is the most iconic mushroom in modern-day popular culture. In many languages, its vernacular names are fly agaric and fly amanita. Indeed, steeped in a bowl of milk, it was used to Citation: Carboué, Q.; Lopez, M. catch flies in houses for centuries in Europe due to its ability to attract and intoxicate flies. Amanita muscaria: Ecology, Chemistry, Although considered poisonous when ingested fresh, this mushroom has been consumed Myths. Encyclopedia 2021, 1, 905–914. as edible in many different places, such as Italy and Mexico [1]. Many traditional recipes https://doi.org/10.3390/ involving boiling the mushroom—the water containing most of the water-soluble toxic encyclopedia1030069 compounds is then discarded—are available. In Japan, the mushroom is dried, soaked in brine for 12 weeks, and rinsed in successive washings before being eaten [2]. -

Inhalant Abuse Pediatric Care
CLINICAL REPORT Guidance for the Clinician in Rendering Inhalant Abuse Pediatric Care Janet F. Williams, MD, Michael Storck, MD, and the Committee on Substance Abuse and Committee on Native American Child Health ABSTRACT Inhalant abuse is the intentional inhalation of a volatile substance for the purpose of achieving an altered mental state. As an important, yet-underrecognized form of substance abuse, inhalant abuse crosses all demographic, ethnic, and socioeco- nomic boundaries, causing significant morbidity and mortality in school-aged and older children. This clinical report reviews key aspects of inhalant abuse, empha- sizes the need for greater awareness, and offers advice regarding the pediatrician’s role in the prevention and management of this substance abuse problem. TYPES OF CHEMICALS AND PRODUCTS ABUSED The term “inhalant” encompasses a wide range of pharmacologically diverse substances that readily vaporize. Most other substances of abuse are classified by grouping together substances that share a specific central nervous system action or perceived psychoactive effect, but inhalant substances that are abused are grouped by having a common route of drug use. Inhalant abuse, sometimes referred to as solvent or volatile substance abuse, can be better understood when the expansive list of inhalants is classified into 3 groups on the basis of what is currently known pharmacologically: group I includes volatile solvents, fuels, and anesthetics; group II includes nitrous oxide; and group III includes volatile alkyl nitrites (Table 1). This classification is also consistent with reported differences in user populations, patterns of abuse, and associated problems seen clinically.1–3 Drugs that do not www.pediatrics.org/cgi/doi/10.1542/ readily vaporize at room temperature, such as cocaine, heroin, nicotine, or alcohol, peds.2007-0470 can also be abused through inhalation, but characteristic pharmacologic properties doi:10.1542/peds.2007-0470 distinguish these substances from inhalants. -

The Effects of the Morphine Analogue Levorphanol on Leukocytes: Metabolic Effects at Rest and During Phagocytosis
The effects of the morphine analogue levorphanol on leukocytes: Metabolic effects at rest and during phagocytosis Nancy Wurster, … , Penelope Pettis, Sharon Lebow J Clin Invest. 1971;50(5):1091-1099. https://doi.org/10.1172/JCI106580. Studies on bacteria have suggested that morphine-like drugs have effects on the cell membrane. To determine the effect of this class of drugs on a mammalian cell, we selected the rabbit peritoneal exudate granulocyte, which undergoes striking membrane changes during phagocytosis. We examined the effect in vitro of the morphine analogue, levorphanol on phagocytosis and metabolism by granulocytes incubated with and without polystyrene particles or live Escherichia coli. Levorphanol (1 or 2 mmoles/liter) decreased: (a) acylation of lysolecithin or lysophosphatidylethanolamine in the medium (which is stimulated about two-fold during phagocytosis) both at rest (40%) and during phagocytosis (60%); (b) uptake of latex particles and Escherichia coli, as judged by electron microscopy; (c) killing of live Escherichia coli (10-fold); (d) 14 14 + CO2 production from glucose-1- C during phagocytosis by at least 80%; (e) K content of granulocytes (35%); (f) oxidation of linoleate-1-14C by 50%, and its incorporation into triglyceride by more than 80%. However, levorphanol stimulated 2 to 3-fold the incorporation of linoleate-1-14C or palmitate-1-14C into several phospholipids. Glucose uptake, lactate production, and adenosine triphosphate (ATP) content are not affected by the drug. Thus, levorphanol does not appear to exert its effects through generalized metabolic suppression. Removal of levorphanol by twice resuspending the granulocytes completely reverses all inhibition. In line with observations on bacteria, it appears that the complex effects of levorphanol on […] Find the latest version: https://jci.me/106580/pdf The Effects of the Morphine Analogue Levorphanol on Leukocytes METABOLIC EFFECTS AT REST AND DURING PHAGOCYTOSIS NANcY WuRsTE, PETER ELSBACH, ERIc J. -

Consultation Documents
WHO Drug Information Vol. 29, No. 3, 2015 Consultation documents To receive draft monographs by email please contact Mrs Wendy Bonny ([email protected]), specifying that you wish to be added to the electronic mailing list. The International Pharmacopoeia Carbamazepinum Carbamazepine This is a draft proposal for The International Pharmacopoeia (Working document QAS/15.608, July 2015). The working document with line numbers and tracked changes is available for comment at www.who.int/medicines/areas/quality_safety/quality_assurance/projects/en/. Please address any comments to: World Health Organization, Quality Assurance and Safety: Medicines, Dr Herbert Schmidt, 1211 Geneva 27, Switzerland; fax: +41 22 791 4730; email: [email protected]. [Note from the Secretariat. It is proposed to revise the monograph on Carbamazepine in The International Pharmacopoeia.] [Note from the editor. In accordance with WHO editorial policy the text reproduced below does not include tracked changes. Changes from the current monograph are indicated by insert and delete in the working document available at the above-mentioned web address.] Molecular formula. C15H12N2O Relative molecular mass. 236.3 Graphic formula. Chemical name. 5H-Dibenz[b,f]azepine-5-carboxamide; CAS Reg. No. 298-46-4. Description. A white to almost white, crystalline powder. Solubility. Practically insoluble in water; sparingly soluble in acetone; soluble in ethanol (~750 g/L) TS; freely soluble in dichloromethane. Category. Antiepileptic. 352 WHO Drug Information Vol. 29, No. 3, 2015 Consultation documents Additional information. Carbamazepine exhibits polymorphism. The acceptable crystalline form is anhydrous polymorph form III1. It corresponds to carbamazepine RS. Storage. Carbamazepine should be kept in a tightly closed container. -

Positron Emission Tomography Studies of Potential Mechanisms Underlying Phencyclidine-Induced Alterations in Striatal Dopamine
Neuropsychopharmacology (2003) 28, 2192–2198 & 2003 Nature Publishing Group All rights reserved 0893-133X/03 $25.00 www.neuropsychopharmacology.org Positron Emission Tomography Studies of Potential Mechanisms Underlying Phencyclidine-Induced Alterations in Striatal Dopamine ,1,2 2 1,2 Wynne K Schiffer* , Jean Logan and Stephen L Dewey 1SUNY Stony Brook, Department of Neurobiology and Behavior, Stony Brook, NY, USA; 2Brookhaven National Laboratory, Chemistry Department, Upton, NY, USA 11 Positron emission tomography (PET), in combination with C-raclopride, was used to examine the effects of phencyclidine (PCP) on dopamine (DA) in the primate striatum. In addition, we explored the hypotheses that GABAergic pathways as well as molecular targets beyond the N-methyl-D-aspartate (NMDA) receptor complex (ie dopamine transporter proteins, DAT) contribute to PCP’s effects. In 11 the first series of experiments, C-raclopride was administered at baseline and 30 min following intravenous PCP administration. In the second series of studies, g-vinyl GABA (GVG) was used to assess whether enhanced GABAergic tone altered NMDA antagonist- induced changes in DA. Animals received an initial PET scan followed by pretreatment with GVG (300 mg/kg), then PCP 30 min prior to 11 a second scan. Finally, we explored the possible contributions of DAT blockade to PCP-induced increases in DA. By examining C- cocaine binding a paradigm in which PCP was coadministered with the radiotracer, we assessed the direct competition between these 11 two compounds for the DAT. At 0.1, 0.5, and 1.0 mg/kg, PCP decreased C-raclopride binding by 2.1, 14.9 7 2.2 and 8.18 7 1.1%, 11 respectively. -

Opiate Receptor Subtype Modulation of Dopaminergic Activity the Effects of Mu, Kappa and Sigma Opiates on the Development of Dopaminergic Supersensitivity
University of Rhode Island DigitalCommons@URI Open Access Dissertations 1986 OPIATE RECEPTOR SUBTYPE MODULATION OF DOPAMINERGIC ACTIVITY THE EFFECTS OF MU, KAPPA AND SIGMA OPIATES ON THE DEVELOPMENT OF DOPAMINERGIC SUPERSENSITIVITY Robert W. Dunn University of Rhode Island Follow this and additional works at: https://digitalcommons.uri.edu/oa_diss Recommended Citation Dunn, Robert W., "OPIATE RECEPTOR SUBTYPE MODULATION OF DOPAMINERGIC ACTIVITY THE EFFECTS OF MU, KAPPA AND SIGMA OPIATES ON THE DEVELOPMENT OF DOPAMINERGIC SUPERSENSITIVITY" (1986). Open Access Dissertations. Paper 147. https://digitalcommons.uri.edu/oa_diss/147 This Dissertation is brought to you for free and open access by DigitalCommons@URI. It has been accepted for inclusion in Open Access Dissertations by an authorized administrator of DigitalCommons@URI. For more information, please contact [email protected]. OPIATE RECEPTOR SUBTYPE MODULATION OF DOPAMINERGIC ACTIVITY THE EFFECTS OF MU, KAPPA AND SIGMA OPIATES ON THE DEVELOPMENT OF DOPAMINERGIC SUPERSENSITIVITY BY ROBERT W. DUNN A DISSERTATION SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN PHARMACOLOGY AND TOXICOLOGY UNIVERSITY OF RHODE ISLAND 1986 DOCTOR OF PHILOSOPHY DISSERTATION OF ROBERT W. DUNN Approved: Dissertation Chairman ~~~~----....-..~~.......... -----~----~~~- Dean of the Graduate School UNIVERSITY OF RHODE ISLAND 1986 ABSTRACT The purpose of this research was to examine the effects of mu (µ), kappa (K) and sigma (o) agents namely, morphine (µ), ethylketo cyclazocine ( K), SKF 10,047 ( o), pentazocine (K, o), cyclazocine (K, o) and the mu antagonists, naloxone and naltrexone on dopamine mediated behaviors and the development of haloperidol-induced dopaminergic supersensitivity (DA-SS) in the mouse. Three behavioral paradigms were utilized which are predictive of mesolimbic and/or striatal dopaminergic effects: locomotor activity (mesol imbic); apomorphine-induced stereotyped behavior (striatal) and apomorphine induced climbing behavior (mesolimbic/striatal).