Synthesis, Characterisation and Development of Novel, Validated Methods for the Detection and Quantification of Diphenidine- Derived New Psychoactive Substances
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Review of Market for Octane Enhancers
May 2000 • NREL/SR-580-28193 Review of Market for Octane Enhancers Final Report J.E. Sinor Consultants, Inc. Niwot, Colorado National Renewable Energy Laboratory 1617 Cole Boulevard Golden, Colorado 80401-3393 NREL is a U.S. Department of Energy Laboratory Operated by Midwest Research Institute • Battelle • Bechtel Contract No. DE-AC36-99-GO10337 May 2000 • NREL/SR-580-28193 Review of Market for Octane Enhancers Final Report J.E. Sinor Consultants, Inc. Niwot, Colorado NREL Technical Monitor: K. Ibsen Prepared under Subcontract No. TXE-0-29113-01 National Renewable Energy Laboratory 1617 Cole Boulevard Golden, Colorado 80401-3393 NREL is a U.S. Department of Energy Laboratory Operated by Midwest Research Institute • Battelle • Bechtel Contract No. DE-AC36-99-GO10337 NOTICE This report was prepared as an account of work sponsored by an agency of the United States government. Neither the United States government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States government or any agency thereof. Available electronically at http://www.doe.gov/bridge Available for a processing fee to U.S. -

Precursors and Chemicals Frequently Used in the Illicit Manufacture of Narcotic Drugs and Psychotropic Substances 2017
INTERNATIONAL NARCOTICS CONTROL BOARD Precursors and chemicals frequently used in the illicit manufacture of narcotic drugs and psychotropic substances 2017 EMBARGO Observe release date: Not to be published or broadcast before Thursday, 1 March 2018, at 1100 hours (CET) UNITED NATIONS CAUTION Reports published by the International Narcotics Control Board in 2017 The Report of the International Narcotics Control Board for 2017 (E/INCB/2017/1) is supplemented by the following reports: Narcotic Drugs: Estimated World Requirements for 2018—Statistics for 2016 (E/INCB/2017/2) Psychotropic Substances: Statistics for 2016—Assessments of Annual Medical and Scientific Requirements for Substances in Schedules II, III and IV of the Convention on Psychotropic Substances of 1971 (E/INCB/2017/3) Precursors and Chemicals Frequently Used in the Illicit Manufacture of Narcotic Drugs and Psychotropic Substances: Report of the International Narcotics Control Board for 2017 on the Implementation of Article 12 of the United Nations Convention against Illicit Traffic in Narcotic Drugs and Psychotropic Substances of 1988 (E/INCB/2017/4) The updated lists of substances under international control, comprising narcotic drugs, psychotropic substances and substances frequently used in the illicit manufacture of narcotic drugs and psychotropic substances, are contained in the latest editions of the annexes to the statistical forms (“Yellow List”, “Green List” and “Red List”), which are also issued by the Board. Contacting the International Narcotics Control Board The secretariat of the Board may be reached at the following address: Vienna International Centre Room E-1339 P.O. Box 500 1400 Vienna Austria In addition, the following may be used to contact the secretariat: Telephone: (+43-1) 26060 Fax: (+43-1) 26060-5867 or 26060-5868 Email: [email protected] The text of the present report is also available on the website of the Board (www.incb.org). -

FSI-D-16-00226R1 Title
Elsevier Editorial System(tm) for Forensic Science International Manuscript Draft Manuscript Number: FSI-D-16-00226R1 Title: An overview of Emerging and New Psychoactive Substances in the United Kingdom Article Type: Review Article Keywords: New Psychoactive Substances Psychostimulants Lefetamine Hallucinogens LSD Derivatives Benzodiazepines Corresponding Author: Prof. Simon Gibbons, Corresponding Author's Institution: UCL School of Pharmacy First Author: Simon Gibbons Order of Authors: Simon Gibbons; Shruti Beharry Abstract: The purpose of this review is to identify emerging or new psychoactive substances (NPS) by undertaking an online survey of the UK NPS market and to gather any data from online drug fora and published literature. Drugs from four main classes of NPS were identified: psychostimulants, dissociative anaesthetics, hallucinogens (phenylalkylamine-based and lysergamide-based materials) and finally benzodiazepines. For inclusion in the review the 'user reviews' on drugs fora were selected based on whether or not the particular NPS of interest was used alone or in combination. NPS that were use alone were considered. Each of the classes contained drugs that are modelled on existing illegal materials and are now covered by the UK New Psychoactive Substances Bill in 2016. Suggested Reviewers: Title Page (with authors and addresses) An overview of Emerging and New Psychoactive Substances in the United Kingdom Shruti Beharry and Simon Gibbons1 Research Department of Pharmaceutical and Biological Chemistry UCL School of Pharmacy -

Organocatalytic Asymmetric N-Sulfonyl Amide C-N Bond Activation to Access Axially Chiral Biaryl Amino Acids
ARTICLE https://doi.org/10.1038/s41467-020-14799-8 OPEN Organocatalytic asymmetric N-sulfonyl amide C-N bond activation to access axially chiral biaryl amino acids Guanjie Wang1, Qianqian Shi2, Wanyao Hu1, Tao Chen1, Yingying Guo1, Zhouli Hu1, Minghua Gong1, ✉ ✉ ✉ Jingcheng Guo1, Donghui Wei 2 , Zhenqian Fu 1,3 & Wei Huang1,3 1234567890():,; Amides are among the most fundamental functional groups and essential structural units, widely used in chemistry, biochemistry and material science. Amide synthesis and trans- formations is a topic of continuous interest in organic chemistry. However, direct catalytic asymmetric activation of amide C-N bonds still remains a long-standing challenge due to high stability of amide linkages. Herein, we describe an organocatalytic asymmetric amide C-N bonds cleavage of N-sulfonyl biaryl lactams under mild conditions, developing a general and practical method for atroposelective construction of axially chiral biaryl amino acids. A structurally diverse set of axially chiral biaryl amino acids are obtained in high yields with excellent enantioselectivities. Moreover, a variety of axially chiral unsymmetrical biaryl organocatalysts are efficiently constructed from the resulting axially chiral biaryl amino acids by our present strategy, and show competitive outcomes in asymmetric reactions. 1 Key Laboratory of Flexible Electronics & Institute of Advanced Materials, Jiangsu National Synergetic Innovation Center for Advanced Materials, Nanjing Tech University, 30 South Puzhu Road, Nanjing 211816, China. 2 College -
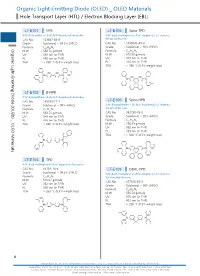
Organic Light-Emitting Diode (OLED) OLED Materials Hole Transport Layer (HTL) / Electron Blocking Layer (EBL)
Organic Light-EmittingOrganic Light-Emitting DiodeHole Transport(OLED) Layer (HTL) / Diode (OLED) _ OLED Materials Electron Blocking Layer (EBL) Organic Light-Emitting Diode (OLED) _ OLED Materials Hole Transport Layer (HTL) / Electron Blocking Layer (EBL) LT-E101 NPB LT-E105 Spiro-TPD N,N'-Bis(naphthalen-1-yl)-N,N'-bis(phenyl)-benzidine N,N'-Bis(3-methylphenyl)-N,N'-bis(phenyl)-2,7-diamino- CAS No. : 123847-85-8 9,9-spirobifluorene Grade : Sublimed, > 99.5% (HPLC) CAS No. : 1033035-83-4 : Grade : Sublimed, > 99% (HPLC) Organic Light-Emitting Diode (OLED) _ OLED Materials Formula C44H32N2 M.W. : 588.74 g/mole Formula : C51H38N2 UV : 339 nm (in THF) M.W. : 678.86 g/mole PL : 450 nm (in THF) UV : 379 nm (in THF) TGA : > 350 °C (0.5% weight loss) PL : 416 nm (in THF) TGA : > 280 °C (0.5% weight loss) N N N N LT-E102 β-NPB N,N'-Bis(naphthalen-2-yl)-N,N'-bis(phenyl)-benzidine Spiro-NPB CAS No. : 139255-17-7 LT-E106 Grade : Sublimed, > 99% (HPLC) N,N'-Bis(naphthalen-1-yl)-N,N'-bis(phenyl)-2,7-diamino- 9,9-spirobifluorene Formula : C44H32N2 M.W. : 588.74 g/mole CAS No. : 932739-76-9 UV : 349 nm (in THF) Grade : Sublimed, > 99% (HPLC) PL : 416 nm (in THF) Formula : C57H38N2 TGA : > 330 °C (0.5% weight loss) M.W. : 750.93 g/mole UV : 380 nm (in THF) PL : 453 nm (in THF) TGA : > 360 °C (0.5% weight loss) N N N N LT-E103 TPD N,N'-Bis(3-methylphenyl)-N,N'-bis(phenyl)-benzidine CAS No. -

Acute Toxicity Associated with the Recreational Use of the Novel Dissociative Psychoactive Substance Methoxphenidine
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2014 Acute toxicity associated with the recreational use of the novel dissociative psychoactive substance methoxphenidine Hofer, K E ; Degrandi, C ; Müller, D M ; Zürrer-Härdi, U ; Wahl, S ; Rauber-Lüthy, C ; Ceschi, A Abstract: INTRODUCTION: Methoxphenidine is a novel dissociative designer drug of the diarylethy- lamine class which shares structural features with phencyclidine (PCP), and is not at present subject to restrictive regulations. There is very limited information about the acute toxicity profile of methoxpheni- dine and the only sources are anonymous internet sites and a 1989 patent of the Searle Company. We report a case of analytically confirmed oral methoxphenidine toxicity. CASE DETAILS: A 53-year-old man was found on the street in a somnolent and confusional state. Observed signs and symptoms such as tachycardia (112 bpm), hypertension (220/125 mmHg), echolalia, confusion, agitation, opisthotonus, nys- tagmus and amnesia were consistent with phencyclidine-induced adverse effects. Temperature (99.1°F (37.3°C)) and peripheral oxygen saturation while breathing room air (99%) were normal. Laboratory analysis revealed an increase of creatine kinase (max 865 U/L), alanine aminotransferase (72 U/L) and gamma-glutamyl transpeptidase (123 U/L). Methoxphenidine was identified by a liquid chromatogra- phy tandem mass spectrometry toxicological screening method using turbulent flow online extraction in plasma and urine samples collected on admission. The clinical course was favourable and signs and symptoms resolved with symptomatic treatment. CONCLUSION: Based on this case report and users’ web reports, and compatible with the chemical structure, methoxphenidine produces effects similar to those of the arylcyclohexylamines, as PCP. -

Synthetic Drugs: Overview and Issues for Congress
Synthetic Drugs: Overview and Issues for Congress Lisa N. Sacco Analyst in Illicit Drugs and Crime Policy Kristin Finklea Specialist in Domestic Security May 3, 2016 Congressional Research Service 7-5700 www.crs.gov R42066 Synthetic Drugs: Overview and Issues for Congress Summary Synthetic drugs, as opposed to natural drugs, are chemically produced in a laboratory. Their chemical structure can be either identical to or different from naturally occurring drugs, and their effects are designed to mimic or even enhance those of natural drugs. When produced clandestinely, they are not typically controlled pharmaceutical substances intended for legitimate medical use. Designer drugs are a form of synthetic drugs. They contain slightly modified molecular structures of illegal or controlled substances, and they are modified in order to circumvent existing drug laws. While the issue of synthetic drugs and their abuse is not new, Congress has demonstrated a renewed concern with the issue. From 2009 to 2011, synthetic drug abuse was reported to have dramatically increased. During this time period, calls to poison control centers for incidents relating to harmful effects of synthetic cannabinoids (such as “K2” and “Spice”) and stimulants (such as “bath salts”) increased at what some considered to be an alarming rate. The number of hospital emergency department visits involving synthetic cannabinoids more than doubled from 2010 to 2011. In 2012 and 2013, however, the number of calls to poison control centers for incidents relating to harmful effects of synthetic cannabinoids and synthetic stimulants decreased. Calls regarding bath salts have declined each year since 2011, while calls regarding synthetic cannabinoids have increased since the drops in 2012 and 2013. -

2020 International Narcotics Control Strategy Report
United States Department of State Bureau for International Narcotics and Law Enforcement Affairs International Narcotics Control Strategy Report Volume I Drug and Chemical Control March 2020 INCSR 2020 Volume 1 Table of Contents Table of Contents Common Abbreviations ..................................................................................................................................... iii International Agreements.................................................................................................................................... v INTRODUCTION ..................................................................................................................................... 1 Legislative Basis for the INCSR ......................................................................................................................... 2 Presidential Determination ................................................................................................................................. 7 Policy and Program Developments .................................................................................................... 12 Overview ......................................................................................................................................................... 13 Methodology for U.S. Government Estimates of Illegal Drug Production .......................................................... 18 Parties to UN Conventions .............................................................................................................................. -

Drug-Related Crime
NT OF ME J T US U.S. Department of Justice R T A I P C E E D B O J C S Office of Justice Programs F A V M F O I N A C IJ S R E BJ G O OJJDP O F PR Bureau of Justice Statistics JUSTICE Drugs & Crime Data September 1994, NCJ–149286 Fact Sheet: Drug-Related Crime Drugs are related to crime in multiple ways. Most This fact sheet will focus on the second and third catego- directly, it is a crime to use, possess, manufacture, or ries. Drug-related offenses and a drug-using lifestyle are distribute drugs classified as having a potential for abuse. major contributors to the U.S. crime problem. Cocaine, heroin, marijuana, and amphetamines are examples of drugs classified to have abuse potential. Drug users in the general population are more Drugs are also related to crime through the effects they likely than nonusers to commit crimes have on the user’s behavior and by generating violence and other illegal activity in connection with drug traffick- The U.S. Department of Health and Human Services ing. The following scheme summarizes the various ways (HHS) National Household Survey on Drug Abuse asks that drugs and crime are related. individuals living in households about their drug and alcohol use and their involvement in acts that could get Summary of drugs/crime relationship them in trouble with the police. Provisional data for 1991 show that among adult respondents (ages 18–49), those Drugs and crime who use cannabis (marijuana) or cocaine were much more relationship Definition Examples likely to commit crimes of all types than those who did Drug-defined Violations of laws Drug possession or offenses prohibiting or reg- use. -

Virginia Acts of Assembly -- 2021 Special Session I
VIRGINIA ACTS OF ASSEMBLY -- 2021 SPECIAL SESSION I CHAPTER 110 An Act to amend and reenact §§ 3.2-4112, 3.2-4113, 3.2-4114.2, 3.2-4115, 3.2-4116, 3.2-4118, 3.2-4119, 18.2-247, 18.2-251.1:3, 54.1-3401, and 54.1-3446 of the Code of Virginia, relating to industrial hemp; emergency. [H 2078] Approved March 12, 2021 Be it enacted by the General Assembly of Virginia: 1. That §§ 3.2-4112, 3.2-4113, 3.2-4114.2, 3.2-4115, 3.2-4116, 3.2-4118, 3.2-4119, 18.2-247, 18.2-251.1:3, 54.1-3401, and 54.1-3446 of the Code of Virginia are amended and reenacted as follows: § 3.2-4112. Definitions. As used in this chapter, unless the context requires a different meaning: "Cannabis sativa product" means a product made from any part of the plant Cannabis sativa, including seeds thereof and any derivative, extract, cannabinoid, isomer, acid, salt, or salt of an isomer, whether growing or not, with a concentration of tetrahydrocannabinol that is greater than that allowed by federal law. "Deal" means to buy temporarily possess industrial hemp grown in compliance with state or federal law and to sell such industrial hemp to a person who that (i) processes industrial hemp in compliance with state or federal law or has not been processed and (ii) sells industrial hemp to a person who processes industrial hemp in compliance with state or federal law was not grown and will not be processed by the person temporarily possessing it. -

The Market for Amphetamine-Type Stimulants and Their Precursors in Oceania
The market for amphetamine-type stimulants and their precursors in Oceania Andreas Schloenhardt Research and Public Policy Series No. 81 The market for amphetamine-type stimulants and their precursors in Oceania Andreas Schloenhardt Research and Public Policy Series No. 81 © Australian Institute of Criminology 2007 ISSN 1326-6004 ISBN 978 1 921185 53 3 Apart from any fair dealing for the purpose of private study, research, criticism or review, as permitted under the Copyright Act 1968 (Cth), no part of this publication may in any form or by any means (electronic, mechanical, microcopying, photocopying, recording or otherwise) be reproduced, stored in a retrieval system or transmitted without prior written permission. Inquiries should be addressed to the publisher. Project no. 0135 Published by the Australian Institute of Criminology GPO Box 2944 Canberra ACT 2601 Tel: (02) 6260 9272 Fax: (02) 6260 9299 Email: [email protected] Website: http://www.aic.gov.au Please note: minor revisions are occasionally made to publications after release. The online versions available on the AIC website will always include any revisions. Edited and typeset by the Australian Institute of Criminology A full list of publications in the Research and Public Policy Series can be found on the Australian Institute of Criminology website at http://www.aic.gov.au Director’s introduction In 2005, the United Nations Office on Drugs and Crime (2005) valued the global market for ATS at US$44 billion, approximately 13.8 percent of the global illicit drug market (at retail level). It has been estimated that Oceania accounts for about 9–10 percent of the global ATS market; a market share that is disproportionately large in relation to the small population of the region. -

Drugs and Crime Facts
U.S. Department of Justice Office of Justice Programs Bureau of Justice Statistics Drugs and Crime Facts By Tina L. Dorsey BJS Editor Priscilla Middleton BJS Digital Information Specialist NCJ 165148 U.S. Department of Justice Office of Justice Programs 810 Seventh Street, N.W. Washington, D.C. 20531 Eric H. Holder, Jr. Attorney General Office of Justice Programs Partnerships for Safer Communities Laurie O. Robinson Acting Assistant Attorney General World Wide Web site: http//www.ojp.usdoj.gov Bureau of Justice Statistics Michael D. Sinclair Acting Director World Wide Web site: http://www.ojp.usdoj.gov/bjs For information contact National Criminal Justice Reference Service 1-800-851-3420 BJS: Bureau of Justice Statistics Drugs and Crime Facts Drugs & Crime Facts This site summarizes U.S. statistics about drug-related crimes, law enforcement, courts, and corrections from Bureau of Justice Statistics (BJS) and non-BJS sources (See Drug data produced by BJS below). It updates the information published in Drugs and Crime Facts, 1994, (NCJ 154043) and will be revised as new information becomes available. The data provide policymakers, criminal justice practitioners, researchers, and the general public with online access to understandable information on various drug law violations and drug-related law enforcement. Contents Drug use and crime Drug law violations Enforcement (arrests, seizures, and operations) Pretrial release, prosecution, and adjudication Correctional populations and facilities Drug treatment under correctional supervision Drug control budget Drug use (by youth and the general population) Public opinion about drugs Bibliography To ease printing, a consolidated version in Adobe Acrobat format (669 KB) of all of the web pages in Drugs & Crime Facts is available for downloading.