Characterization of Inositol Transporters As a Method for Drug Delivery to the Central Nervous System
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

To Study Mutant P53 Gain of Function, Various Tumor-Derived P53 Mutants
Differential effects of mutant TAp63γ on transactivation of p53 and/or p63 responsive genes and their effects on global gene expression. A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science By Shama K Khokhar M.Sc., Bilaspur University, 2004 B.Sc., Bhopal University, 2002 2007 1 COPYRIGHT SHAMA K KHOKHAR 2007 2 WRIGHT STATE UNIVERSITY SCHOOL OF GRADUATE STUDIES Date of Defense: 12-03-07 I HEREBY RECOMMEND THAT THE THESIS PREPARED UNDER MY SUPERVISION BY SHAMA KHAN KHOKHAR ENTITLED Differential effects of mutant TAp63γ on transactivation of p53 and/or p63 responsive genes and their effects on global gene expression BE ACCEPTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Master of Science Madhavi P. Kadakia, Ph.D. Thesis Director Daniel Organisciak , Ph.D. Department Chair Committee on Final Examination Madhavi P. Kadakia, Ph.D. Steven J. Berberich, Ph.D. Michael Leffak, Ph.D. Joseph F. Thomas, Jr., Ph.D. Dean, School of Graduate Studies 3 Abstract Khokhar, Shama K. M.S., Department of Biochemistry and Molecular Biology, Wright State University, 2007 Differential effect of TAp63γ mutants on transactivation of p53 and/or p63 responsive genes and their effects on global gene expression. p63, a member of the p53 gene family, known to play a role in development, has more recently also been implicated in cancer progression. Mice lacking p63 exhibit severe developmental defects such as limb truncations, abnormal skin, and absence of hair follicles, teeth, and mammary glands. Germline missense mutations of p63 have been shown to be responsible for several human developmental syndromes including SHFM, EEC and ADULT syndromes and are associated with anomalies in the development of organs of epithelial origin. -

The Concise Guide to Pharmacology 2019/20
Edinburgh Research Explorer THE CONCISE GUIDE TO PHARMACOLOGY 2019/20 Citation for published version: Cgtp Collaborators 2019, 'THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters', British Journal of Pharmacology, vol. 176 Suppl 1, pp. S397-S493. https://doi.org/10.1111/bph.14753 Digital Object Identifier (DOI): 10.1111/bph.14753 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: British Journal of Pharmacology General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 28. Sep. 2021 S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Transporters. British Journal of Pharmacology (2019) 176, S397–S493 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters Stephen PH Alexander1 , Eamonn Kelly2, Alistair Mathie3 ,JohnAPeters4 , Emma L Veale3 , Jane F Armstrong5 , Elena Faccenda5 ,SimonDHarding5 ,AdamJPawson5 , Joanna L -
And Mir183 in Mir183/96 Dko Mutant Mice (Top) And
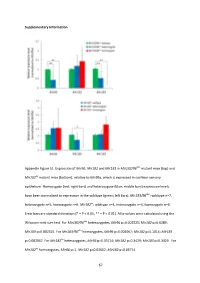
Supplementary Information Appendix Figure S1. Expression of Mir96 , Mir182 and Mir183 in Mir183/96 dko mutant mice (top) and Mir182 ko mutant mice (bottom), relative to Mir99a , which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96 dko : wildtype n=7, heterozygote n=5, homozygote n=6. Mir182 ko : wildtype n=4, heterozygote n=4, homozygote n=4. Error bars are standard deviation (* = P < 0.05, ** = P < 0.01). All p-values were calculated using the Wilcoxon rank sum test. For Mir183/96 dko heterozygotes, Mir96 p=0.002525; Mir182 p=0.6389; Mir183 p=0.002525. For Mir183/96 dko homozygotes, Mir96 p=0.002067; Mir182 p=0.1014; Mir183 p=0.002067. For Mir182 ko heterozygotes, Mir96 p=0.05714; Mir182 p=0.3429; Mir183 p=0.3429. For Mir182 ko homozygotes, Mir96 p=1; Mir182 p=0.02652; Mir183 p=0.05714. 67 68 Appendix Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96 dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 69 70 Appendix Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182 ko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 71 Appendix Figure S4. Mean ABR waveforms at 12kHz, shown at 20dB (top) and 50dB (bottom) above threshold (sensation level, SL) ± standard deviation, at four weeks old. -

Transport of Sugars
BI84CH32-Frommer ARI 29 April 2015 12:34 Transport of Sugars Li-Qing Chen,1,∗ Lily S. Cheung,1,∗ Liang Feng,3 Widmar Tanner,2 and Wolf B. Frommer1 1Department of Plant Biology, Carnegie Institution for Science, Stanford, California 94305; email: [email protected] 2Zellbiologie und Pflanzenbiochemie, Universitat¨ Regensburg, 93040 Regensburg, Germany 3Department of Molecular and Cellular Physiology, Stanford University School of Medicine, Stanford, California 94305 Annu. Rev. Biochem. 2015. 84:865–94 Keywords First published online as a Review in Advance on glucose, sucrose, carrier, GLUT, SGLT, SWEET March 5, 2015 The Annual Review of Biochemistry is online at Abstract biochem.annualreviews.org Soluble sugars serve five main purposes in multicellular organisms: as sources This article’s doi: of carbon skeletons, osmolytes, signals, and transient energy storage and as 10.1146/annurev-biochem-060614-033904 transport molecules. Most sugars are derived from photosynthetic organ- Copyright c 2015 by Annual Reviews. isms, particularly plants. In multicellular organisms, some cells specialize All rights reserved in providing sugars to other cells (e.g., intestinal and liver cells in animals, ∗ These authors contributed equally to this review. photosynthetic cells in plants), whereas others depend completely on an ex- Annu. Rev. Biochem. 2015.84:865-894. Downloaded from www.annualreviews.org ternal supply (e.g., brain cells, roots and seeds). This cellular exchange of Access provided by b-on: Universidade de Lisboa (UL) on 09/05/16. For personal use only. sugars requires transport proteins to mediate uptake or release from cells or subcellular compartments. Thus, not surprisingly, sugar transport is criti- cal for plants, animals, and humans. -

HOXB6 Homeo Box B6 HOXB5 Homeo Box B5 WNT5A Wingless-Type
5 6 6 5 . 4 2 1 1 1 2 4 6 4 3 2 9 9 7 0 5 7 5 8 6 4 0 8 2 3 1 8 3 7 1 0 0 4 0 2 5 0 8 7 5 4 1 1 0 3 6 0 4 8 3 7 4 7 6 9 6 7 1 5 0 8 1 4 1 1 7 1 0 0 4 2 0 8 1 1 1 2 5 3 5 0 7 2 6 9 1 2 1 8 3 5 2 9 8 0 6 0 9 5 1 9 9 2 1 1 6 0 2 3 0 3 6 9 1 6 5 5 7 1 1 2 1 1 7 5 4 6 6 4 1 1 2 8 4 7 1 6 2 7 7 5 4 3 2 4 3 6 9 4 1 7 1 3 4 1 2 1 3 1 1 4 7 3 1 1 1 1 5 3 2 6 1 5 1 3 5 4 5 2 3 1 1 6 1 7 3 2 5 4 3 1 6 1 5 3 1 7 6 5 1 1 1 4 6 1 6 2 7 2 1 2 e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a a S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S S HOXB6 homeo box B6 HOXB5 homeo box B5 WNT5A wingless-type MMTV integration site family, member 5A WNT5A wingless-type MMTV integration site family, member 5A FKBP11 FK506 binding protein 11, 19 kDa EPOR erythropoietin receptor SLC5A6 solute carrier family 5 sodium-dependent vitamin transporter, member 6 SLC5A6 solute carrier family 5 sodium-dependent vitamin transporter, member 6 RAD52 RAD52 homolog S. -

(12) Patent Application Publication (10) Pub. No.: US 2003/0082511 A1 Brown Et Al
US 20030082511A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2003/0082511 A1 Brown et al. (43) Pub. Date: May 1, 2003 (54) IDENTIFICATION OF MODULATORY Publication Classification MOLECULES USING INDUCIBLE PROMOTERS (51) Int. Cl." ............................... C12O 1/00; C12O 1/68 (52) U.S. Cl. ..................................................... 435/4; 435/6 (76) Inventors: Steven J. Brown, San Diego, CA (US); Damien J. Dunnington, San Diego, CA (US); Imran Clark, San Diego, CA (57) ABSTRACT (US) Correspondence Address: Methods for identifying an ion channel modulator, a target David B. Waller & Associates membrane receptor modulator molecule, and other modula 5677 Oberlin Drive tory molecules are disclosed, as well as cells and vectors for Suit 214 use in those methods. A polynucleotide encoding target is San Diego, CA 92121 (US) provided in a cell under control of an inducible promoter, and candidate modulatory molecules are contacted with the (21) Appl. No.: 09/965,201 cell after induction of the promoter to ascertain whether a change in a measurable physiological parameter occurs as a (22) Filed: Sep. 25, 2001 result of the candidate modulatory molecule. Patent Application Publication May 1, 2003 Sheet 1 of 8 US 2003/0082511 A1 KCNC1 cDNA F.G. 1 Patent Application Publication May 1, 2003 Sheet 2 of 8 US 2003/0082511 A1 49 - -9 G C EH H EH N t R M h so as se W M M MP N FIG.2 Patent Application Publication May 1, 2003 Sheet 3 of 8 US 2003/0082511 A1 FG. 3 Patent Application Publication May 1, 2003 Sheet 4 of 8 US 2003/0082511 A1 KCNC1 ITREXCHO KC 150 mM KC 2000000 so 100 mM induced Uninduced Steady state O 100 200 300 400 500 600 700 Time (seconds) FIG. -

Supplemental Table 3 Site ID Intron Poly(A) Site Type NM/KG Inum
Supplemental Table 3 Site ID Intron Poly(A) site Type NM/KG Inum Region Gene ID Gene Symbol Gene Annotation Hs.120277.1.10 chr3:170997234:170996860 170996950 b NM_153353 7 CDS 151827 LRRC34 leucine rich repeat containing 34 Hs.134470.1.27 chr17:53059664:53084458 53065543 b NM_138962 10 CDS 124540 MSI2 musashi homolog 2 (Drosophila) Hs.162889.1.18 chr14:80367239:80329208 80366262 b NM_152446 12 CDS 145508 C14orf145 chromosome 14 open reading frame 145 Hs.187898.1.27 chr22:28403623:28415294 28404458 b NM_181832 16 3UTR 4771 NF2 neurofibromin 2 (bilateral acoustic neuroma) Hs.228320.1.6 chr10:115527009:115530350 115527470 b BC036365 5 CDS 79949 C10orf81 chromosome 10 open reading frame 81 Hs.266308.1.2 chr11:117279579:117278191 117278967 b NM_032046 12 CDS 84000 TMPRSS13 transmembrane protease, serine 13 Hs.266308.1.4 chr11:117284536:117281662 117283722 b NM_032046 9 CDS 84000 TMPRSS13 transmembrane protease, serine 13 Hs.2689.1.4 chr10:53492398:53563605 53492622 b NM_006258 7 CDS 5592 PRKG1 protein kinase, cGMP-dependent, type I Hs.280781.1.6 chr18:64715646:64829150 64715837 b NM_024781 4 CDS 79839 C18orf14 chromosome 18 open reading frame 14 Hs.305985.2.25 chr12:8983686:8984438 8983942 b BX640639 17 3UTR NA NA NA Hs.312098.1.36 chr1:151843991:151844258 151844232 b NM_003815 15 CDS 8751 ADAM15 a disintegrin and metalloproteinase domain 15 (metargidin) Hs.314338.1.11 chr21:39490293:39481214 39487623 b NM_018963 41 CDS 54014 BRWD1 bromodomain and WD repeat domain containing 1 Hs.33368.1.3 chr15:92685158:92689361 92688314 b NM_018349 6 CDS 55784 MCTP2 multiple C2-domains with two transmembrane regions 2 Hs.346736.1.21 chr2:99270738:99281614 99272414 b AK126402 10 3UTR 51263 MRPL30 mitochondrial ribosomal protein L30 Hs.445061.1.19 chr16:69322898:69290216 69322712 b NM_018052 14 CDS 55697 VAC14 Vac14 homolog (S. -

Supplementary Table 2
Supplementary Table 2. Differentially Expressed Genes following Sham treatment relative to Untreated Controls Fold Change Accession Name Symbol 3 h 12 h NM_013121 CD28 antigen Cd28 12.82 BG665360 FMS-like tyrosine kinase 1 Flt1 9.63 NM_012701 Adrenergic receptor, beta 1 Adrb1 8.24 0.46 U20796 Nuclear receptor subfamily 1, group D, member 2 Nr1d2 7.22 NM_017116 Calpain 2 Capn2 6.41 BE097282 Guanine nucleotide binding protein, alpha 12 Gna12 6.21 NM_053328 Basic helix-loop-helix domain containing, class B2 Bhlhb2 5.79 NM_053831 Guanylate cyclase 2f Gucy2f 5.71 AW251703 Tumor necrosis factor receptor superfamily, member 12a Tnfrsf12a 5.57 NM_021691 Twist homolog 2 (Drosophila) Twist2 5.42 NM_133550 Fc receptor, IgE, low affinity II, alpha polypeptide Fcer2a 4.93 NM_031120 Signal sequence receptor, gamma Ssr3 4.84 NM_053544 Secreted frizzled-related protein 4 Sfrp4 4.73 NM_053910 Pleckstrin homology, Sec7 and coiled/coil domains 1 Pscd1 4.69 BE113233 Suppressor of cytokine signaling 2 Socs2 4.68 NM_053949 Potassium voltage-gated channel, subfamily H (eag- Kcnh2 4.60 related), member 2 NM_017305 Glutamate cysteine ligase, modifier subunit Gclm 4.59 NM_017309 Protein phospatase 3, regulatory subunit B, alpha Ppp3r1 4.54 isoform,type 1 NM_012765 5-hydroxytryptamine (serotonin) receptor 2C Htr2c 4.46 NM_017218 V-erb-b2 erythroblastic leukemia viral oncogene homolog Erbb3 4.42 3 (avian) AW918369 Zinc finger protein 191 Zfp191 4.38 NM_031034 Guanine nucleotide binding protein, alpha 12 Gna12 4.38 NM_017020 Interleukin 6 receptor Il6r 4.37 AJ002942 -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

An NKX2-1/ERK/WNT Feedback Loop Modulates Gastric Identity And
RESEARCH ARTICLE An NKX2-1/ERK/WNT feedback loop modulates gastric identity and response to targeted therapy in lung adenocarcinoma Rediet Zewdu1,2, Elnaz Mirzaei Mehrabad1,3, Kelley Ingram1,4, Pengshu Fang1,4, Katherine L Gillis1,4, Soledad A Camolotto1,2, Grace Orstad1,4, Alex Jones1,2, Michelle C Mendoza1,4, Benjamin T Spike1,4, Eric L Snyder1,2,4* 1Huntsman Cancer Institute, Salt Lake City, United States; 2Department of Pathology, University of Utah, Salt Lake City, United States; 3School of Computing, University of Utah, Salt Lake City, United States; 4Department of Oncological Sciences, University of Utah, Salt Lake City, United States Abstract Cancer cells undergo lineage switching during natural progression and in response to therapy. NKX2-1 loss in human and murine lung adenocarcinoma leads to invasive mucinous adenocarcinoma (IMA), a lung cancer subtype that exhibits gastric differentiation and harbors a distinct spectrum of driver oncogenes. In murine BRAFV600E-driven lung adenocarcinoma, NKX2-1 is required for early tumorigenesis, but dispensable for established tumor growth. NKX2-1-deficient, BRAFV600E-driven tumors resemble human IMA and exhibit a distinct response to BRAF/MEK inhibitors. Whereas BRAF/MEK inhibitors drive NKX2-1-positive tumor cells into quiescence, NKX2- 1-negative cells fail to exit the cell cycle after the same therapy. BRAF/MEK inhibitors induce cell identity switching in NKX2-1-negative lung tumors within the gastric lineage, which is driven in part by WNT signaling and FoxA1/2. These data elucidate a complex, reciprocal relationship between lineage specifiers and oncogenic signaling pathways in the regulation of lung adenocarcinoma identity that is likely to impact lineage-specific therapeutic strategies. -

Strand Breaks for P53 Exon 6 and 8 Among Different Time Course of Folate Depletion Or Repletion in the Rectosigmoid Mucosa
SUPPLEMENTAL FIGURE COLON p53 EXONIC STRAND BREAKS DURING FOLATE DEPLETION-REPLETION INTERVENTION Supplemental Figure Legend Strand breaks for p53 exon 6 and 8 among different time course of folate depletion or repletion in the rectosigmoid mucosa. The input of DNA was controlled by GAPDH. The data is shown as ΔCt after normalized to GAPDH. The higher ΔCt the more strand breaks. The P value is shown in the figure. SUPPLEMENT S1 Genes that were significantly UPREGULATED after folate intervention (by unadjusted paired t-test), list is sorted by P value Gene Symbol Nucleotide P VALUE Description OLFM4 NM_006418 0.0000 Homo sapiens differentially expressed in hematopoietic lineages (GW112) mRNA. FMR1NB NM_152578 0.0000 Homo sapiens hypothetical protein FLJ25736 (FLJ25736) mRNA. IFI6 NM_002038 0.0001 Homo sapiens interferon alpha-inducible protein (clone IFI-6-16) (G1P3) transcript variant 1 mRNA. Homo sapiens UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 15 GALNTL5 NM_145292 0.0001 (GALNT15) mRNA. STIM2 NM_020860 0.0001 Homo sapiens stromal interaction molecule 2 (STIM2) mRNA. ZNF645 NM_152577 0.0002 Homo sapiens hypothetical protein FLJ25735 (FLJ25735) mRNA. ATP12A NM_001676 0.0002 Homo sapiens ATPase H+/K+ transporting nongastric alpha polypeptide (ATP12A) mRNA. U1SNRNPBP NM_007020 0.0003 Homo sapiens U1-snRNP binding protein homolog (U1SNRNPBP) transcript variant 1 mRNA. RNF125 NM_017831 0.0004 Homo sapiens ring finger protein 125 (RNF125) mRNA. FMNL1 NM_005892 0.0004 Homo sapiens formin-like (FMNL) mRNA. ISG15 NM_005101 0.0005 Homo sapiens interferon alpha-inducible protein (clone IFI-15K) (G1P2) mRNA. SLC6A14 NM_007231 0.0005 Homo sapiens solute carrier family 6 (neurotransmitter transporter) member 14 (SLC6A14) mRNA. -

To Nucleotide Sequence Determinations (Pancreatic Dnase I/U1 Ribonuclease/U2 Ribonuclease/Pancreatic Ribonuclease A/Ribosubstituted DNA) GARY V
Proc. Nat. Acad. Sci. USA Vol. 71, No. 12, pp. 5017-5021, December 1974 Deoxysubstitution in RNA by RNA Polymerase In Vitro: A New Approach to Nucleotide Sequence Determinations (pancreatic DNase I/U1 ribonuclease/U2 ribonuclease/pancreatic ribonuclease A/ribosubstituted DNA) GARY V. PADDOCK, HOWARD C. HEINDELL AND WINSTON SALSER Biology Department and Molecular Biology Institute, University of California at Los Angeles, 405 Hilgard Ave., Los Angeles, Calif. 90024 Communicated by Charles Yanofsky, August 12, 1974 ABSTRACT Deoxynucleotides have been incorporated substituted RNA. We have observed that Mn++ ion will not into RNA synthesized in vitro by RNA polymerase with only cause DNA polymerase to synthesize ribosubstituted either double-stranded or single-stranded DNA as a to synthesize template. By use of this technique to block or promote DNA but will also cause RNA polymerase cleavage at a particular phosphodiester bond, a variety of deoxysubstituted RNA. Subsequently we have discovered a specific cleavages may be obtained with the available ribo- number of other papers dealing with the synthesis of deoxy- nucleases and deoxyribonuclease I. These methods should substituted RNA (12-14). These papers did not, however, greatly increase the ease and rapidity of nucleotide se- point out the applicability of the approach to nucleotide quence determinations. sequencing and did not include the controls we have found The introduction of radiographic approaches by Sanger and essential for demonstrating that complete deoxysubstitution his colleagues (1-4) greatly increased the power of nucleotide has occurred. sequencing techniques, but there remain certain obstacles In this preliminary paper we discuss the theory by which whose solution could result in impressive further increases in substitution of deoxynucleotides becomes an aid in nucleotide the rapidity with which large sequences may be determined.