Clinical, Molecular and Genetic Aspects
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Expression Polarization
Transcriptional Profiling of the Human Monocyte-to-Macrophage Differentiation and Polarization: New Molecules and Patterns of Gene Expression This information is current as of September 27, 2021. Fernando O. Martinez, Siamon Gordon, Massimo Locati and Alberto Mantovani J Immunol 2006; 177:7303-7311; ; doi: 10.4049/jimmunol.177.10.7303 http://www.jimmunol.org/content/177/10/7303 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2006/11/03/177.10.7303.DC1 Material http://www.jimmunol.org/ References This article cites 61 articles, 22 of which you can access for free at: http://www.jimmunol.org/content/177/10/7303.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 27, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2006 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Transcriptional Profiling of the Human Monocyte-to-Macrophage Differentiation and Polarization: New Molecules and Patterns of Gene Expression1 Fernando O. -

Small Cell Ovarian Carcinoma: Genomic Stability and Responsiveness to Therapeutics
Gamwell et al. Orphanet Journal of Rare Diseases 2013, 8:33 http://www.ojrd.com/content/8/1/33 RESEARCH Open Access Small cell ovarian carcinoma: genomic stability and responsiveness to therapeutics Lisa F Gamwell1,2, Karen Gambaro3, Maria Merziotis2, Colleen Crane2, Suzanna L Arcand4, Valerie Bourada1,2, Christopher Davis2, Jeremy A Squire6, David G Huntsman7,8, Patricia N Tonin3,4,5 and Barbara C Vanderhyden1,2* Abstract Background: The biology of small cell ovarian carcinoma of the hypercalcemic type (SCCOHT), which is a rare and aggressive form of ovarian cancer, is poorly understood. Tumourigenicity, in vitro growth characteristics, genetic and genomic anomalies, and sensitivity to standard and novel chemotherapeutic treatments were investigated in the unique SCCOHT cell line, BIN-67, to provide further insight in the biology of this rare type of ovarian cancer. Method: The tumourigenic potential of BIN-67 cells was determined and the tumours formed in a xenograft model was compared to human SCCOHT. DNA sequencing, spectral karyotyping and high density SNP array analysis was performed. The sensitivity of the BIN-67 cells to standard chemotherapeutic agents and to vesicular stomatitis virus (VSV) and the JX-594 vaccinia virus was tested. Results: BIN-67 cells were capable of forming spheroids in hanging drop cultures. When xenografted into immunodeficient mice, BIN-67 cells developed into tumours that reflected the hypercalcemia and histology of human SCCOHT, notably intense expression of WT-1 and vimentin, and lack of expression of inhibin. Somatic mutations in TP53 and the most common activating mutations in KRAS and BRAF were not found in BIN-67 cells by DNA sequencing. -
![SLC2A6 Mouse Monoclonal Antibody [Clone ID: OTI7E3] Product Data](https://docslib.b-cdn.net/cover/9408/slc2a6-mouse-monoclonal-antibody-clone-id-oti7e3-product-data-59408.webp)
SLC2A6 Mouse Monoclonal Antibody [Clone ID: OTI7E3] Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA500639 SLC2A6 Mouse Monoclonal Antibody [Clone ID: OTI7E3] Product data: Product Type: Primary Antibodies Clone Name: OTI7E3 Applications: IF, IHC, WB Recommend Dilution: IHC 1:200, WB: 1:200 Reactivity: Human Host: Mouse Isotype: IgG2a Clonality: Monoclonal Immunogen: Full-length protein expressed in 293T cell transfected with human SLC2A6 expression vector Formulation: PBS (pH 7.3) containing 1% BSA, 50% glycerol and 0.02% sodium azide. Concentration: 1.47 mg/ml Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Predicted Protein Size: 54.5 kDa Gene Name: solute carrier family 2 member 6 Database Link: NP_060055 Entrez Gene 11182 Human Background: Hexose transport into mammalian cells is catalyzed by a family of membrane proteins, including SLC2A6, that contain 12 transmembrane domains and a number of critical conserved residues. Synonyms: GLUT6; GLUT9; HSA011372 Protein Families: Transmembrane This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2020 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 4 SLC2A6 Mouse Monoclonal Antibody [Clone ID: OTI7E3] – TA500639 Product images: HEK293T cells were transfected with the pCMV6- ENTRY control (Left lane) or pCMV6-ENTRY SLC2A6 ([RC204391], Right lane) cDNA for 48 hrs and lysed. Equivalent amounts of cell lysates (5 ug per lane) were separated by SDS-PAGE and immunoblotted with anti-SLC2A6. -

Viewed Under 23 (B) Or 203 (C) fi M M Male Cko Mice, and Largely Unaffected Magni Cation; Scale Bars, 500 M (B) and 50 M (C)
BRIEF COMMUNICATION www.jasn.org Renal Fanconi Syndrome and Hypophosphatemic Rickets in the Absence of Xenotropic and Polytropic Retroviral Receptor in the Nephron Camille Ansermet,* Matthias B. Moor,* Gabriel Centeno,* Muriel Auberson,* † † ‡ Dorothy Zhang Hu, Roland Baron, Svetlana Nikolaeva,* Barbara Haenzi,* | Natalya Katanaeva,* Ivan Gautschi,* Vladimir Katanaev,*§ Samuel Rotman, Robert Koesters,¶ †† Laurent Schild,* Sylvain Pradervand,** Olivier Bonny,* and Dmitri Firsov* BRIEF COMMUNICATION *Department of Pharmacology and Toxicology and **Genomic Technologies Facility, University of Lausanne, Lausanne, Switzerland; †Department of Oral Medicine, Infection, and Immunity, Harvard School of Dental Medicine, Boston, Massachusetts; ‡Institute of Evolutionary Physiology and Biochemistry, St. Petersburg, Russia; §School of Biomedicine, Far Eastern Federal University, Vladivostok, Russia; |Services of Pathology and ††Nephrology, Department of Medicine, University Hospital of Lausanne, Lausanne, Switzerland; and ¶Université Pierre et Marie Curie, Paris, France ABSTRACT Tight control of extracellular and intracellular inorganic phosphate (Pi) levels is crit- leaves.4 Most recently, Legati et al. have ical to most biochemical and physiologic processes. Urinary Pi is freely filtered at the shown an association between genetic kidney glomerulus and is reabsorbed in the renal tubule by the action of the apical polymorphisms in Xpr1 and primary fa- sodium-dependent phosphate transporters, NaPi-IIa/NaPi-IIc/Pit2. However, the milial brain calcification disorder.5 How- molecular identity of the protein(s) participating in the basolateral Pi efflux remains ever, the role of XPR1 in the maintenance unknown. Evidence has suggested that xenotropic and polytropic retroviral recep- of Pi homeostasis remains unknown. Here, tor 1 (XPR1) might be involved in this process. Here, we show that conditional in- we addressed this issue in mice deficient for activation of Xpr1 in the renal tubule in mice resulted in impaired renal Pi Xpr1 in the nephron. -

Two Locus Inheritance of Non-Syndromic Midline Craniosynostosis Via Rare SMAD6 and 4 Common BMP2 Alleles 5 6 Andrew T
1 2 3 Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and 4 common BMP2 alleles 5 6 Andrew T. Timberlake1-3, Jungmin Choi1,2, Samir Zaidi1,2, Qiongshi Lu4, Carol Nelson- 7 Williams1,2, Eric D. Brooks3, Kaya Bilguvar1,5, Irina Tikhonova5, Shrikant Mane1,5, Jenny F. 8 Yang3, Rajendra Sawh-Martinez3, Sarah Persing3, Elizabeth G. Zellner3, Erin Loring1,2,5, Carolyn 9 Chuang3, Amy Galm6, Peter W. Hashim3, Derek M. Steinbacher3, Michael L. DiLuna7, Charles 10 C. Duncan7, Kevin A. Pelphrey8, Hongyu Zhao4, John A. Persing3, Richard P. Lifton1,2,5,9 11 12 1Department of Genetics, Yale University School of Medicine, New Haven, CT, USA 13 2Howard Hughes Medical Institute, Yale University School of Medicine, New Haven, CT, USA 14 3Section of Plastic and Reconstructive Surgery, Department of Surgery, Yale University School of Medicine, New Haven, CT, USA 15 4Department of Biostatistics, Yale University School of Medicine, New Haven, CT, USA 16 5Yale Center for Genome Analysis, New Haven, CT, USA 17 6Craniosynostosis and Positional Plagiocephaly Support, New York, NY, USA 18 7Department of Neurosurgery, Yale University School of Medicine, New Haven, CT, USA 19 8Child Study Center, Yale University School of Medicine, New Haven, CT, USA 20 9The Rockefeller University, New York, NY, USA 21 22 ABSTRACT 23 Premature fusion of the cranial sutures (craniosynostosis), affecting 1 in 2,000 24 newborns, is treated surgically in infancy to prevent adverse neurologic outcomes. To 25 identify mutations contributing to common non-syndromic midline (sagittal and metopic) 26 craniosynostosis, we performed exome sequencing of 132 parent-offspring trios and 59 27 additional probands. -

Regulation of Skeletal Muscle Glucose Transport and Glucose Metabolism by Exercise Training
nutrients Review Regulation of Skeletal Muscle Glucose Transport and Glucose Metabolism by Exercise Training Parker L. Evans 1,2,3, Shawna L. McMillin 1,2,3 , Luke A. Weyrauch 1,2,3 and Carol A. Witczak 1,2,3,4,* 1 Department of Kinesiology, East Carolina University, Greenville, NC 27858, USA; [email protected] (P.L.E.); [email protected] (S.L.M.); [email protected] (L.A.W.) 2 Department of Physiology, Brody School of Medicine, East Carolina University, Greenville, NC 27834, USA 3 East Carolina Diabetes & Obesity Institute, East Carolina University, Greenville, NC 27834, USA 4 Department of Biochemistry & Molecular Biology, Brody School of Medicine, East Carolina University, Greenville, NC 27834, USA * Correspondence: [email protected]; Tel.: +1-252-744-1224 Received: 8 September 2019; Accepted: 8 October 2019; Published: 12 October 2019 Abstract: Aerobic exercise training and resistance exercise training are both well-known for their ability to improve human health; especially in individuals with type 2 diabetes. However, there are critical differences between these two main forms of exercise training and the adaptations that they induce in the body that may account for their beneficial effects. This article reviews the literature and highlights key gaps in our current understanding of the effects of aerobic and resistance exercise training on the regulation of systemic glucose homeostasis, skeletal muscle glucose transport and skeletal muscle glucose metabolism. Keywords: aerobic exercise; blood glucose; functional overload; GLUT; hexokinase; insulin resistance; resistance exercise; SGLT; type 2 diabetes; weightlifting 1. Introduction Exercise training is defined as planned bouts of physical activity which repeatedly occur over a duration of time lasting from weeks to years. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Gene Expression Patterns and Tumor Uptake of 18F-FDG, 18F-FLT, and 18F-FEC in PET/MRI of an Orthotopic Mouse Xenotransplantation Model of Pancreatic Cancer
Journal of Nuclear Medicine, published on July 16, 2008 as doi:10.2967/jnumed.107.050021 Gene Expression Patterns and Tumor Uptake of 18F-FDG, 18F-FLT, and 18F-FEC in PET/MRI of an Orthotopic Mouse Xenotransplantation Model of Pancreatic Cancer Corinna von Forstner*1, Jan-Hendrik Egberts*2, Ole Ammerpohl2, Dagmara Niedzielska3, Ralph Buchert3, Pal Mikecz3, Udo Schumacher4, Kersten Peldschus5, Gerhard Adam5, Christian Pilarsky6, Robert Grutzmann6, Holger Kalthoff2, Eberhard Henze1, and Winfried Brenner3 1Department of Nuclear Medicine, University Hospital of Schleswig-Holstein, Campus Kiel, Kiel, Germany; 2Department of General Surgery and Thoracic Surgery, University Hospital of Schleswig-Holstein, Campus Kiel, Kiel, Germany; 3Department of Nuclear Medicine, University Medical Center Hamburg-Eppendorf, Hamburg, Germany; 4Department of Anatomy II Experimental Morphology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany; 5Department of Diagnostic and Interventional Radiology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany; and 6Department of Visceral, Thoracic, and Vascular Surgery, University Hospital Carl Gustav Carus, Technical University of Dresden, Dresden, Germany the highest and most consistent uptake in various human pan- Our aim was to use PET/MRI to evaluate and compare the uptake creatic tumor cell lines in SCID mice. The imaging results could of 18F-FDG, 3-deoxy-3-18F-fluorothymidine (18F-FLT), and 18F- be explained by gene expression patterns of membrane trans- fluorethylcholine (18F-FEC) in human pancreatic tumor cell lines porters and enzymes for tracer uptake and retention as mea- after xenotransplantation into SCID mice and to correlate tumor sured by gene array analysis and quantitative polymerase uptake with gene expression of membrane transporters and chain reaction in the respective cell lines. -

Protein Identities in Evs Isolated from U87-MG GBM Cells As Determined by NG LC-MS/MS
Protein identities in EVs isolated from U87-MG GBM cells as determined by NG LC-MS/MS. No. Accession Description Σ Coverage Σ# Proteins Σ# Unique Peptides Σ# Peptides Σ# PSMs # AAs MW [kDa] calc. pI 1 A8MS94 Putative golgin subfamily A member 2-like protein 5 OS=Homo sapiens PE=5 SV=2 - [GG2L5_HUMAN] 100 1 1 7 88 110 12,03704523 5,681152344 2 P60660 Myosin light polypeptide 6 OS=Homo sapiens GN=MYL6 PE=1 SV=2 - [MYL6_HUMAN] 100 3 5 17 173 151 16,91913397 4,652832031 3 Q6ZYL4 General transcription factor IIH subunit 5 OS=Homo sapiens GN=GTF2H5 PE=1 SV=1 - [TF2H5_HUMAN] 98,59 1 1 4 13 71 8,048185945 4,652832031 4 P60709 Actin, cytoplasmic 1 OS=Homo sapiens GN=ACTB PE=1 SV=1 - [ACTB_HUMAN] 97,6 5 5 35 917 375 41,70973209 5,478027344 5 P13489 Ribonuclease inhibitor OS=Homo sapiens GN=RNH1 PE=1 SV=2 - [RINI_HUMAN] 96,75 1 12 37 173 461 49,94108966 4,817871094 6 P09382 Galectin-1 OS=Homo sapiens GN=LGALS1 PE=1 SV=2 - [LEG1_HUMAN] 96,3 1 7 14 283 135 14,70620005 5,503417969 7 P60174 Triosephosphate isomerase OS=Homo sapiens GN=TPI1 PE=1 SV=3 - [TPIS_HUMAN] 95,1 3 16 25 375 286 30,77169764 5,922363281 8 P04406 Glyceraldehyde-3-phosphate dehydrogenase OS=Homo sapiens GN=GAPDH PE=1 SV=3 - [G3P_HUMAN] 94,63 2 13 31 509 335 36,03039959 8,455566406 9 Q15185 Prostaglandin E synthase 3 OS=Homo sapiens GN=PTGES3 PE=1 SV=1 - [TEBP_HUMAN] 93,13 1 5 12 74 160 18,68541938 4,538574219 10 P09417 Dihydropteridine reductase OS=Homo sapiens GN=QDPR PE=1 SV=2 - [DHPR_HUMAN] 93,03 1 1 17 69 244 25,77302971 7,371582031 11 P01911 HLA class II histocompatibility antigen, -

The Concise Guide to Pharmacology 2019/20
Edinburgh Research Explorer THE CONCISE GUIDE TO PHARMACOLOGY 2019/20 Citation for published version: Cgtp Collaborators 2019, 'THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters', British Journal of Pharmacology, vol. 176 Suppl 1, pp. S397-S493. https://doi.org/10.1111/bph.14753 Digital Object Identifier (DOI): 10.1111/bph.14753 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: British Journal of Pharmacology General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 28. Sep. 2021 S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Transporters. British Journal of Pharmacology (2019) 176, S397–S493 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters Stephen PH Alexander1 , Eamonn Kelly2, Alistair Mathie3 ,JohnAPeters4 , Emma L Veale3 , Jane F Armstrong5 , Elena Faccenda5 ,SimonDHarding5 ,AdamJPawson5 , Joanna L -
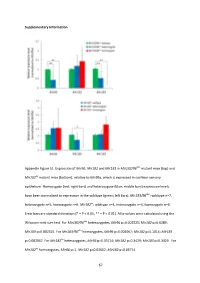
And Mir183 in Mir183/96 Dko Mutant Mice (Top) And
Supplementary Information Appendix Figure S1. Expression of Mir96 , Mir182 and Mir183 in Mir183/96 dko mutant mice (top) and Mir182 ko mutant mice (bottom), relative to Mir99a , which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96 dko : wildtype n=7, heterozygote n=5, homozygote n=6. Mir182 ko : wildtype n=4, heterozygote n=4, homozygote n=4. Error bars are standard deviation (* = P < 0.05, ** = P < 0.01). All p-values were calculated using the Wilcoxon rank sum test. For Mir183/96 dko heterozygotes, Mir96 p=0.002525; Mir182 p=0.6389; Mir183 p=0.002525. For Mir183/96 dko homozygotes, Mir96 p=0.002067; Mir182 p=0.1014; Mir183 p=0.002067. For Mir182 ko heterozygotes, Mir96 p=0.05714; Mir182 p=0.3429; Mir183 p=0.3429. For Mir182 ko homozygotes, Mir96 p=1; Mir182 p=0.02652; Mir183 p=0.05714. 67 68 Appendix Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96 dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 69 70 Appendix Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182 ko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 71 Appendix Figure S4. Mean ABR waveforms at 12kHz, shown at 20dB (top) and 50dB (bottom) above threshold (sensation level, SL) ± standard deviation, at four weeks old. -

Transport of Sugars
BI84CH32-Frommer ARI 29 April 2015 12:34 Transport of Sugars Li-Qing Chen,1,∗ Lily S. Cheung,1,∗ Liang Feng,3 Widmar Tanner,2 and Wolf B. Frommer1 1Department of Plant Biology, Carnegie Institution for Science, Stanford, California 94305; email: [email protected] 2Zellbiologie und Pflanzenbiochemie, Universitat¨ Regensburg, 93040 Regensburg, Germany 3Department of Molecular and Cellular Physiology, Stanford University School of Medicine, Stanford, California 94305 Annu. Rev. Biochem. 2015. 84:865–94 Keywords First published online as a Review in Advance on glucose, sucrose, carrier, GLUT, SGLT, SWEET March 5, 2015 The Annual Review of Biochemistry is online at Abstract biochem.annualreviews.org Soluble sugars serve five main purposes in multicellular organisms: as sources This article’s doi: of carbon skeletons, osmolytes, signals, and transient energy storage and as 10.1146/annurev-biochem-060614-033904 transport molecules. Most sugars are derived from photosynthetic organ- Copyright c 2015 by Annual Reviews. isms, particularly plants. In multicellular organisms, some cells specialize All rights reserved in providing sugars to other cells (e.g., intestinal and liver cells in animals, ∗ These authors contributed equally to this review. photosynthetic cells in plants), whereas others depend completely on an ex- Annu. Rev. Biochem. 2015.84:865-894. Downloaded from www.annualreviews.org ternal supply (e.g., brain cells, roots and seeds). This cellular exchange of Access provided by b-on: Universidade de Lisboa (UL) on 09/05/16. For personal use only. sugars requires transport proteins to mediate uptake or release from cells or subcellular compartments. Thus, not surprisingly, sugar transport is criti- cal for plants, animals, and humans.