A Network-Based Analysis of Traditional Chinese Medicine Cold and Hot Patterns in Rheumatoid Arthritis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Next-Generation Sequencing of Representational Difference Analysis
Planta DOI 10.1007/s00425-017-2657-0 ORIGINAL ARTICLE Next-generation sequencing of representational difference analysis products for identification of genes involved in diosgenin biosynthesis in fenugreek (Trigonella foenum-graecum) 1 1 1 1 Joanna Ciura • Magdalena Szeliga • Michalina Grzesik • Mirosław Tyrka Received: 19 August 2016 / Accepted: 30 January 2017 Ó The Author(s) 2017. This article is published with open access at Springerlink.com Abstract Within the transcripts related to sterol and steroidal Main conclusion Representational difference analysis saponin biosynthesis, we discovered novel candidate of cDNA was performed and differential products were genes of diosgenin biosynthesis and validated their sequenced and annotated. Candidate genes involved in expression using quantitative RT-PCR analysis. Based on biosynthesis of diosgenin in fenugreek were identified. these findings, we supported the idea that diosgenin is Detailed mechanism of diosgenin synthesis was biosynthesized from cycloartenol via cholesterol. This is proposed. the first report on the next-generation sequencing of cDNA-RDA products. Analysis of the transcriptomes Fenugreek (Trigonella foenum-graecum L.) is a valuable enriched in low copy sequences contributed substantially medicinal and crop plant. It belongs to Fabaceae family to our understanding of the biochemical pathways of and has a unique potential to synthesize valuable steroidal steroid synthesis in fenugreek. saponins, e.g., diosgenin. Elicitation (methyl jasmonate) and precursor feeding (cholesterol and squalene) were Keywords Diosgenin Á Next-generation sequencing Á used to enhance the content of sterols and steroidal Phytosterols Á Representational difference analysis of sapogenins in in vitro grown plants for representational cDNA Á Steroidal saponins Á Transcriptome user-friendly difference analysis of cDNA (cDNA-RDA). -

Relating Metatranscriptomic Profiles to the Micropollutant
1 Relating Metatranscriptomic Profiles to the 2 Micropollutant Biotransformation Potential of 3 Complex Microbial Communities 4 5 Supporting Information 6 7 Stefan Achermann,1,2 Cresten B. Mansfeldt,1 Marcel Müller,1,3 David R. Johnson,1 Kathrin 8 Fenner*,1,2,4 9 1Eawag, Swiss Federal Institute of Aquatic Science and Technology, 8600 Dübendorf, 10 Switzerland. 2Institute of Biogeochemistry and Pollutant Dynamics, ETH Zürich, 8092 11 Zürich, Switzerland. 3Institute of Atmospheric and Climate Science, ETH Zürich, 8092 12 Zürich, Switzerland. 4Department of Chemistry, University of Zürich, 8057 Zürich, 13 Switzerland. 14 *Corresponding author (email: [email protected] ) 15 S.A and C.B.M contributed equally to this work. 16 17 18 19 20 21 This supporting information (SI) is organized in 4 sections (S1-S4) with a total of 10 pages and 22 comprises 7 figures (Figure S1-S7) and 4 tables (Table S1-S4). 23 24 25 S1 26 S1 Data normalization 27 28 29 30 Figure S1. Relative fractions of gene transcripts originating from eukaryotes and bacteria. 31 32 33 Table S1. Relative standard deviation (RSD) for commonly used reference genes across all 34 samples (n=12). EC number mean fraction bacteria (%) RSD (%) RSD bacteria (%) RSD eukaryotes (%) 2.7.7.6 (RNAP) 80 16 6 nda 5.99.1.2 (DNA topoisomerase) 90 11 9 nda 5.99.1.3 (DNA gyrase) 92 16 10 nda 1.2.1.12 (GAPDH) 37 39 6 32 35 and indicates not determined. 36 37 38 39 S2 40 S2 Nitrile hydration 41 42 43 44 Figure S2: Pearson correlation coefficients r for rate constants of bromoxynil and acetamiprid with 45 gene transcripts of ECs describing nucleophilic reactions of water with nitriles. -

( 12 ) United States Patent
US010208322B2 (12 ) United States Patent ( 10 ) Patent No. : US 10 ,208 , 322 B2 Coelho et al. (45 ) Date of Patent: * Feb . 19, 2019 ( 54 ) IN VIVO AND IN VITRO OLEFIN ( 56 ) References Cited CYCLOPROPANATION CATALYZED BY HEME ENZYMES U . S . PATENT DOCUMENTS 3 , 965 ,204 A 6 / 1976 Lukas et al. (71 ) Applicant: California Institute of Technology , 4 , 243 ,819 A 1 / 1981 Henrick Pasadena , CA (US ) 5 ,296 , 595 A 3 / 1994 Doyle 5 , 703 , 246 A 12 / 1997 Aggarwal et al. 7 , 226 , 768 B2 6 / 2007 Farinas et al. ( 72 ) Inventors : Pedro S . Coelho , Los Angeles, CA 7 , 267 , 949 B2 9 / 2007 Richards et al . (US ) ; Eric M . Brustad , Durham , NC 7 ,625 ,642 B2 12 / 2009 Matsutani et al. (US ) ; Frances H . Arnold , La Canada , 7 ,662 , 969 B2 2 / 2010 Doyle et al. CA (US ) ; Zhan Wang , San Jose , CA 7 ,863 ,030 B2 1 / 2011 Arnold (US ) ; Jared C . Lewis , Chicago , IL 8 ,247 ,430 B2 8 / 2012 Yuan 8 , 993 , 262 B2 * 3 / 2015 Coelho . .. .. .. • * • C12P 7 /62 (US ) 435 / 119 9 ,399 , 762 B26 / 2016 Farwell et al . (73 ) Assignee : California Institute of Technology , 9 , 493 ,799 B2 * 11 /2016 Coelho .. C12P 7162 Pasadena , CA (US ) 2006 / 0030718 AL 2 / 2006 Zhang et al. 2006 / 0111347 A1 5 / 2006 Askew , Jr . et al. 2007 /0276013 AL 11 /2007 Ebbinghaus et al . ( * ) Notice : Subject to any disclaimer , the term of this 2009 /0238790 A2 9 /2009 Sommadosi et al. patent is extended or adjusted under 35 2010 / 0056806 A1 3 / 2010 Warren U . -

Geneid T-Test Q-Val Gene Symbol Gene Title GO Biological Process
Supplementary Table 2. Genes selected by FADA as the most discriminant between tumoral and normal prostate samples GO Biological Process GO Molecular Function GO Cellular Component GeneID T-test q-val Gene Symbol Gene Title Description Description Description Pathway serine-type endopeptidase inhibitor activity /// endopeptidase inhibitor serpin peptidase inhibitor, clade activity /// serine-type endopeptidase 213572_s_at -6.753 2.213E-05 SERPINB1 B (ovalbumin), member 1 --- inhibitor activity cytoplasm --- ion transport /// cellular defense response /// positive regulation of T-cell, immune regulator 1, cell proliferation /// proton plasma membrane /// integral to ATPase, H+ transporting, transport /// transport /// proton transporter activity /// hydrogen ion plasma membrane /// membrane /// 204158_s_at -5.729 8.585E-05 TCIRG1 lysosomal V0 subunit A3 transport transporter activity integral to membrane --- chemotaxis /// cell adhesion /// homophilic cell adhesion /// nervous system development /// cell differentiation /// positive roundabout, axon guidance regulation of axonogenesis /// receptor activity /// axon guidance integral to plasma membrane /// cell receptor, homolog 1 development /// nervous system receptor activity /// identical protein surface /// membrane /// integral to 213194_at -6.168 4.441E-05 ROBO1 (Drosophila) development binding /// protein binding membrane --- structural molecule activity /// nucleus /// intermediate filament /// protein binding /// microtubule cytoplasmic dynein complex /// 203411_s_at -5.297 1.636E-04 -

SUPPLEMENTARY MATERIAL Supplementary Table 1
Predicting Drug Promiscuity Using Spherical Harmonic Surface The Open Conference Proceedings Journal, 2011, Volume 2 i SUPPLEMENTARY MATERIAL Supplementary Table 1. MDDR Target Annotations Used in the Promiscuity Predictions TARGET_KEY EXTERNAL_ID 1001 prostaglandin-endoperoxide synthase 1002 cyclooxygenase 1 1003 cyclooxygenase 2 1046 ribonucleoside-diphosphate reductase 1112 thymidylate synthase 1209 phosphoribosylglycinamide formyltransferase 1609 purine-nucleoside phosphorylase 1611 uridine phosphorylase 1637 nad adp-ribosyltransferase 1657 transferring alkyl or aryl groups, other than methyl groups 1798 thymidine kinase 1814 protein kinase 1843 1-phosphatidylinositol 4-kinase 1883 protein-tyrosine kinase 1973 nucleotidyltransferase 2018 rna-directed dna polymerase 2121 phospholipase a2 2124 acetylcholinesterase 2282 phosphoric diester hydrolase 2283 phosphodiesterase i 2309 3',5'-cyclic-nucleotide phosphodiesterase 2310 3',5'-cyclic-gmp phosphodiesterase 2316 steryl-sulfatase 2414 alpha-glucosidase 2565 adenosylhomocysteinase 2576 peptidase 25 aldehyde reductase 2613 dipeptidyl-peptidase iv 2619 peptidyl-dipeptidase a 2624 serine-type d-ala-d-ala carboxypeptidase 2659 serine endopeptidase 2663 trypsin 2664 thrombin 2665 coagulation factor xa 2672 coagulation factor viia 2695 tryptase 2742 cathepsin b 2750 cathepsin l 2765 caspase-1 2784 hiv-1 retropepsin 2811 metalloendopeptidase 2822 stromelysin 1 286 xanthine oxidase 2978 beta-lactamase 3014 adenosine deaminase ii The Open Conference Proceedings Journal, 2011, Volume 2 Pérez-Nueno -

Dalmanol Biosyntheses Require Coupling of Two Separate Polyketide Gene Clusters
Showcasing research progress from Professor Ren Xiang Tan’s As featured in: laboratories at Nanjing University & Nanjing University of Chinese Medicine, Nanjing, China. Dalmanol biosyntheses require coupling of two separate polyketide gene clusters The biosynthesis of immunosuppressants (dalmanol A and acetodalmanol A) is characterized by the coupling of two separate (naphthalene- and chromane-encoding) gene clusters under the catalysis of unspecifi c enzyme(s) that may distribute widely in the fungal kingdom. The fi nding highlights an access to the inter-cluster orchestration derived chemodiversity of microbial secondary metabolites that remain to be a reliable source of leading compounds triggering the development of See Ren Xiang Tan et al., new medicines, agrochemicals, and other tool molecules. Chem. Sci., 2019, 10, 73. rsc.li/chemical-science Registered charity number: 207890 Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Dalmanol biosyntheses require coupling of two separate polyketide gene clusters† Cite this: Chem. Sci.,2019,10,73 a a bc a a All publication charges for this article Zhen Zhen Zhou, Hong Jie Zhu, Li Ping Lin, Xuan Zhang, Hui Ming Ge, have been paid for by the Royal Society Rui Hua Jiaoa and Ren Xiang Tan *ab of Chemistry Polyketide–polyketide hybrids are unique natural products with promising bioactivity, but the hybridization processes remain poorly understood. Herein, we present that the biosynthetic pathways of two immunosuppressants, dalmanol A and acetodalmanol A, result from an unspecific monooxygenase triggered hybridization of two distinct polyketide (naphthalene and chromane) biosynthetic gene clusters. The orchestration of the functional dimorphism of the polyketide synthase (ChrA) ketoreductase (KR) domain (shortened as ChrA KR) with that of the KR partner (ChrB) in the bioassembly line increases the polyketide diversity and allows the fungal generation of plant chromanes (e.g., noreugenin) and phloroglucinols (e.g., 2,4,6-trihydroxyacetophenone). -

Abuse-Deterrent Pharmaceutical Compositions
(19) TZZ¥ _Z¥Z_T (11) EP 3 210 630 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 30.08.2017 Bulletin 2017/35 A61K 47/36 (2006.01) A61K 9/14 (2006.01) A61K 9/16 (2006.01) A61K 9/20 (2006.01) (2006.01) (2006.01) (21) Application number: 16157803.4 A61K 31/455 A61K 31/485 (22) Date of filing: 29.02.2016 (84) Designated Contracting States: (71) Applicant: G.L. Pharma GmbH AL AT BE BG CH CY CZ DE DK EE ES FI FR GB 8502 Lannach (AT) GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR (72) Inventor: The designation of the inventor has not Designated Extension States: yet been filed BA ME Designated Validation States: (74) Representative: Sonn & Partner Patentanwälte MA MD Riemergasse 14 1010 Wien (AT) (54) ABUSE-DETERRENT PHARMACEUTICAL COMPOSITIONS (57) Disclosed are pharmaceutical compositions comprising oxycodone and an oxycodone-processing enzyme, wherein oxycodone is contained in the pharmaceutical composition in a storage stable, enzyme-reactive state and under conditions wherein no enzymatic activity acts on oxycodone. EP 3 210 630 A1 Printed by Jouve, 75001 PARIS (FR) EP 3 210 630 A1 Description [0001] The present application relates to abuse-deterrent pharmaceutical compositions comprising oxycodone. [0002] Prescription opioid products are an important component of modern pain management. However, abuse and 5 misuse of these products have created a serious and growing public health problem. One potentially important step towards the goal of creating safer opioid analgesics has been the development of opioids that are formulated to deter abuse. -

Tumor-Normal (Log2) Gene Title 1559057 at -1.71 Chrx Orf45 205833 S at -1.73 Prostate Androgen-Regulated Transcript 1 223150 S A
Tumor-Normal (log2) Gene Title 1559057_at -1.71 ChrX orf45 205833_s_at -1.73 Prostate androgen-regulated transcript 1 223150_s_at 0.98 protein tyrosine phosphatase, non-receptor type 23 204856_at 0.91 UDP-GlcNAc:betaGal beta-1,3-NAcGlctransferase 3 232932_at -1.44 Hypothetical protein LOC645030 237669_at -1.13 Protein tyrosine phosphatase domain containing 1 235041_at 0.81 golgi SNAP receptor complex member 2 243580_at 1.16 Guanine nucleotide binding protein (G protein), alpha 14 234354_x_at -1.28 erb-b2 (Her2) erythroblastic leukemia viral oncogene homolog 2 217228_s_at 1.07 ankyrin repeat and SOCS box-containing 4 232690_at -1.53 Mannosyl (alpha-1,3-)-glycoprotein 244371_at 1.36 Hypothetical protein MGC16169 204924_at 1.15 toll-like receptor 2 1559382_at 0.89 chr19 orf 42 239965_at 0.86 hypothetical protein LOC151878 202409_at 4.11 chr11 orf 43 235630_at 0.66 Transcribed locus 236879_at 0.79 Ubiquitin-activating enzyme E1-like 2 221237_s_at -1.59 oxysterol binding protein 2 230443_at -1.30 NHP2 non-histone chromosome protein 2-like 1 208250_s_at -0.70 deleted in malignant brain tumors 1 235814_at -1.18 Transcribed locus 231544_s_at -1.76 CDNA: FLJ21837 fis, clone HEP01664 243767_at 0.83 Transcribed locus 241865_at 0.72 G protein, beta polypeptide 1 237350_at -1.54 similar to CDNA sequence BC021608 227670_at -1.12 zinc finger protein 75a 217100_s_at 0.75 UBX domain containing 7 240422_at -1.19 Flavin containing monooxygenase 5 230000_at 0.87 chr17 orf 27 230599_at 0.80 Ring finger protein 19 204433_s_at -1.18 spermatogenesis associated -
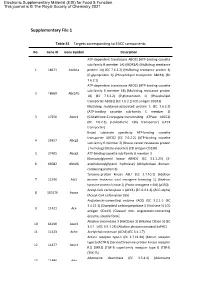
Supplementary File 1
Electronic Supplementary Material (ESI) for Food & Function. This journal is © The Royal Society of Chemistry 2021 Supplementary File 1 Table S1 Targets corresponding to EACC components No. Gene ID Gene Symbol Description ATP-dependent translocase ABCB1 (ATP-binding cassette sub-family B member 1A) (MDR1A) (Multidrug resistance 1 18671 Abcb1a protein 1A) (EC 7.6.2.2) (Multidrug resistance protein 3) (P-glycoprotein 3) (Phospholipid transporter ABCB1) (EC 7.6.2.1) ATP-dependent translocase ABCB1 (ATP-binding cassette sub-family B member 1B) (Multidrug resistance protein 2 18669 Abcb1b 1B) (EC 7.6.2.2) (P-glycoprotein 1) (Phospholipid transporter ABCB1) (EC 7.6.2.1) (CD antigen CD243) Multidrug resistance-associated protein 1 (EC 7.6.2.2) (ATP-binding cassette sub-family C member 1) 3 17250 Abcc1 (Glutathione-S-conjugate-translocating ATPase ABCC1) (EC 7.6.2.3) (Leukotriene C(4) transporter) (LTC4 transporter) Broad substrate specificity ATP-binding cassette transporter ABCG2 (EC 7.6.2.2) (ATP-binding cassette 4 26357 Abcg2 sub-family G member 2) (Breast cancer resistance protein 1 homolog) (Urate exporter) (CD antigen CD338) 5 27405 Abcg3 ATP-binding cassette sub-family G member 3 Monoacylglycerol lipase ABHD6 (EC 3.1.1.23) (2- 6 66082 Abhd6 arachidonoylglycerol hydrolase) (Abhydrolase domain- containing protein 6) Tyrosine-protein kinase ABL1 (EC 2.7.10.2) (Abelson 7 11350 Abl1 murine leukemia viral oncogene homolog 1) (Abelson tyrosine-protein kinase 1) (Proto-oncogene c-Abl) (p150) Acetyl-CoA carboxylase 1 (ACC1) (EC 6.4.1.2) (ACC-alpha) -

Alterations in Progesterone Catabolic Enzymes by Insulin
Graduate Theses, Dissertations, and Problem Reports 2007 Alterations in progesterone catabolic enzymes by insulin Caleb Owens Lemley West Virginia University Follow this and additional works at: https://researchrepository.wvu.edu/etd Recommended Citation Lemley, Caleb Owens, "Alterations in progesterone catabolic enzymes by insulin" (2007). Graduate Theses, Dissertations, and Problem Reports. 2536. https://researchrepository.wvu.edu/etd/2536 This Thesis is protected by copyright and/or related rights. It has been brought to you by the The Research Repository @ WVU with permission from the rights-holder(s). You are free to use this Thesis in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you must obtain permission from the rights-holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/ or on the work itself. This Thesis has been accepted for inclusion in WVU Graduate Theses, Dissertations, and Problem Reports collection by an authorized administrator of The Research Repository @ WVU. For more information, please contact [email protected]. Alterations in Progesterone Catabolic Enzymes by Insulin Caleb Owens Lemley Thesis submitted to the Davis College of Agriculture, Forestry and Consumer Sciences at West Virginia University in partial fulfillment of the requirements for the degree of Master of Science in Reproductive Physiology Matthew E. Wilson, Ph.D., Chair Kenneth P. Blemings, Ph.D. K. Marie Krause, Ph.D. Division of Animal and Nutritional Sciences Morgantown, WV 2007 Keywords: Progesterone Catabolism, Insulin, Glucagon, Cytochrome P450 ABSTRACT Alterations in Progesterone Catabolic Enzymes by Insulin Caleb Owens Lemley The current experiments were conducted to determine if insulin could alter the progesterone catabolic enzymes, cytochrome P450 2C and 3A in vitro, utilizing a mouse hepatocyte cell-line, or in vivo, utilizing the ovariectomized ewe as a model. -

(12) United States Patent (10) Patent No.: US 8,993,262 B2 Coelho Et Al
USOO8993262B2 (12) United States Patent (10) Patent No.: US 8,993,262 B2 Coelho et al. (45) Date of Patent: Mar. 31, 2015 (54) IN VIVO AND IN VITRO OLEFIN 2006/0030718 A1* 2/2006 Zhang et al. .................. 548,954 CYCLOPROPANATION CATALYZED BY 2006/01 11347 A1 5/2006 Askew, Jr. et al. 2007/0276013 A1 1 1/2007 Ebbinghaus et al. HEMIE ENZYMES 2009/0238790 A2 9/2009 Sommadossi et al. 2010.00568.06 A1 3/2010 Warren (71) Applicant: California Institute of Technology, 2010/0168463 A1 7, 2010 Hirata et al. Pasadena, CA (US) 2010/024010.6 A1 9/2010 Wong et al. 2011 0196086 A1 8, 2011 Matsushita et al. (72) Inventors: Pedro S. Coelho, Los Angeles, CA (US); 2012/0237591 A1 9, 2012 Cullis et al. Eric M. Brustad, Durham, NC (US); Frances H. Arnold, La Canada, CA FOREIGN PATENT DOCUMENTS (US); Zhan Wang. San Jose, CA (US); EP O 200 638 B1 4, 1986 Jared C. Lewis, Chicago, IL (US) WO 2007 144599 A2 12/2007 WO 2011, 1595.50 A3 12/2011 (73) Assignee: California Institute of Technology, Pasadena, CA (US) OTHER PUBLICATIONS (*) Notice: Subject to any disclaimer, the term of this Adams, P.D. et al., “PHENIX: a comprehensive Python-based system patent is extended or adjusted under 35 for macromolecular structure solution.” Acta Crystallogr., Sect. D, U.S.C. 154(b) by 0 days. R.JKumar, F.K.R. et R.O.al., 1soprenoid painwayNavonimation optimization Ioror taxoto (21) Appl. No.: 14/185.861 precursor overproduction in Escherichia coli,” Science, 2010, y x- - - 9 330:70-74. -

Supplementary Information for a Proactive Genotype-To-Patient-Phenotype Map for Cystathionine Beta-Synthase
Supplementary information for A proactive genotype-to-patient-phenotype map for cystathionine beta-synthase Song Sun, Jochen Weile, Marta Verby, Atina G. Cote, Yingzhou Wu, Iosifina Fotiadou, Julia Kitaygorodsky, Jasper Rine, Pavel Ješina, Viktor Kožich, Frederick P. Roth Supplementary Figures Supplementary Figure 1. Expression of human CBS protein can rescue growth of yeast cys4D strain in the absence of exogenous cysteine. Growth of cys4D strain expressing CBS gene or carrying empty vector in non-selective (a) or selective (b) medium. The nonselective medium is synthetic complete medium lacking cysteine and supplemented with glutathione, a stable source of cysteine. The selective medium is synthetic complete medium lacking cysteine and glutathione. All medium was supplemented with galactose as carbon source to induce the expression. 1 Supplementary Figure 2. VE map fitness scores are in good agreement with relative growth rates determined in single-variant assays and strong correlation between VE map fitness scores with low concentrations of vitamin B6. (a-c) Correlation between fitness scores from VE maps and relative growth rates from single-variant assays at three different vitamin B6 concentrations. (d) Correlation between fitness scores from high vitamin B6 VE map and Cys-/Cys+ growth ratios from single-variant assays. (e) Correlation between VE map fitness scores with 0 and 1 ng/ml vitamin B6. The correlation test is Pearson correlation. 2 Supplementary Figure 3. VE maps for CBS before computational imputation and refinement: fitness landscape with low level of vitamin B6 (top), fitness landscape with high level of vitamin B6 (middle) and delta fitness (high-low vitamin B6) landscape (bottom).