Fluorine Absorption Coefficients in Superphosphate Den Gases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fluorosilicic Acid CAS No
Product Safety Summary Fluorosilicic Acid CAS No. 16961-83-4 This Product Safety Summary is intended to provide a general overview of the chemical substance. The information in the summary is basic information and is not intended to provide emergency response information, medical information or treatment information. The summary should not be used to provide in-depth safety and health information. In-depth safety and health information can be found in the Safety Data Sheet (SDS) for the chemical substance. Names Fluorosilicic acid (FSA) Hexafluorosilicic acid (HFA or HFS) Sand acid Silicate(2-), hexafluoro- hydrogen (1:2) Silicofluoride Fluosilicic acid Hydroflurosilicic acid Silicofluoric acid Silicon hexafluoride dihydride Silicate(2-), hexafluoro-, dihydrogen Product Overview Solvay Fluorides, LLC does not sell fluorosilicic acid solutions directly to consumers. Most fluorosilicic acid is used in industrial or municipal applications/processes. Concentrated fluorosilicic acid solution (FSA) is used for water fluoridation, as a metal surface treatment and cleaner and for pH adjustment in industrial textile processing or laundries. It can also be used in the processing of hides, for hardening masonry and ceramics and in the manufacture of other chemicals. FSA can only exist as a liquid. There is no solid form. Fluorosilicic acid solutions are corrosive and contact can severely irritate and burn the skin and eyes causing possible permanent eye damage. Breathing concentrated fluorosilicic acid solutions can severely irritate and burn the nose, throat, and lungs, causing nosebleeds, cough, wheezing and shortness of breath. Many of the symptoms described are due to the hydrogen fluoride present as an impurity. Page 1 of 7 Copyright 2010-2013, Solvay America, Inc. -

PRODUCTION of ELECTRONIC GRADE LUNAR SILICON by DISPROPORTIONATION of SILICON DIFLUORIDE, William N
PRODUCTION OF ELECTRONIC GRADE LUNAR SILICON BY DISPROPORTIONATION OF SILICON DIFLUORIDE, William N. Agosto, Lunar Industries, P.O. Box 590004, Houston, TX 77259-0004 Waldron (1) has proposed to extract lunar silicon by sodium reduction of sodium fluorosilicate derived from reacting sodium fluoride with lunar silicon tetrafluoride. Silicon tetrafluoride is obtained by the action of hydrofluoric acid on lunar silicates. While these reactions are well understood, the resulting lunar silicon is not likely to meet electronic specifications of 5 nines purity. Dale and Margrave (2) have shown that silicon difluoride can be obtained by the action of silicon tetrafluoride on elemental silicon at elevated temperatures (1100-1200 C) and low pressures (1-2 torr). The resulting silicon difluoride will then spontaneously disproportionate into hyperpure silicon and silicon tetrafluoride in vacuum at approximately 400 C. On its own merits, silicon difluoride polymerizes into a tough waxy solid in the temperature range from liquid nitrogen to about 100 C. It is the silicon analog of teflon. Silicon difluoride ignites in moist air but is stable under lunar surface conditions and may prove to be a valuable industrial material that is largely lunar derived for lunar surface applications. The most effective driver for lunar industrialization may be the prospects for industrial space solar power systems in orbit or on the moon that are built with lunar materials. Such systems would require large quantities of electronic grade silicon or compound semiconductors for photovoltaics and electronic controls. Since silicon is the most abundant semimetal in the silicate portion of any solar system rock (approximately 20 wt%), lunar silicon production is bound to be an important process in such a solar power project. -

United States Patent Office Patented Apr
3,128,297 United States Patent Office Patented Apr. 7, 1964 1. 2 The various operable organo-silicon compounds, operable 3,128,297 catalysts and reaction conditions will now be defined in PROCESS FOR SELICON-HALOGEN BOND more detail. The term "halogen' as used herein includes RED STRIBUTION the elements fluorine, chlorine, bromine and iodine. Bernard Kanner, Tonawanda, and Donald L. Bailey, Sny Monomeric silanes which can be employed as reactants der, N.Y., assignors to Union Carbide Corporation, a in the process of this invention may be represented by corporation of New York No Drawing. Filed Mar. 31, 1961, Ser. No. 99,667 the formula: 20 Claims. (C. 260-448.2) (B) R (Y-R-) six This invention relates to a process for the redistribution 10 wherein R is a divalent hydrocarbon group, the Y group of silicon-halogen chemical bonds. More particularly, is hydrogen, fluoro, chloro, bromo, iodo, cyano, the invention is directed to a process for the redistribution O of silicon-fluorine and other silicon-halogen bonds, pref -COO G, -NG, G -O G, erably silicon-chlorine bonds, in organo-silicon com -V-va, pounds. This application is a continuation-in-part of our 5 or nitro, the R' group is hydrogen, the vinyl group or an copending application Serial No. 15,841, filed March 18, Y-R- group, X is a halogen, G is a monovalent hy 1960, now abandoned. drocarbon group, e is an integer having a value from 0 We have discovered that an efficient and rapid redis to 3, f is an integer having a value from 0 to 1 and the tribution of silicon-fluorine and other silicon-halogen sum of e and f is never greater than 3. -

Silicon Tetrafluoride on Io
Icarus, in press Silicon Tetrafluoride on Io Laura Schaefer and Bruce Fegley, Jr. Planetary Chemistry Laboratory, Department of Earth and Planetary Sciences, Washington University, St. Louis, MO 63130-4899 [email protected] [email protected] ABSTRACT Silicon tetrafluoride (SiF4) is observed in terrestrial volcanic gases and is predicted to be the major F – bearing species in low temperature volcanic gases on Io (Schaefer and Fegley, 2005b). SiF4 gas is also a potential indicator of silica – rich crust on Io. We used F/S ratios in terrestrial and extraterrestrial basalts, and gas/lava enrichment factors for F and S measured at terrestrial volcanoes to calculate equilibrium SiF4/SO2 ratios in volcanic gases on Io. We conclude that SiF4 can be produced at levels comparable to the observed NaCl/SO2 gas ratio. We also considered potential loss processes for SiF4 in volcanic plumes and in Io’s atmosphere including ion-molecule reactions, electron chemistry, photochemistry, reactions with the major atmospheric constituents, and condensation. Photochemical destruction (tchem ~266 days) and/or condensation as Na2SiF6 (s) appear to be the major sinks for SiF4. We recommend searching for SiF4 with infrared spectroscopy using its 9.7 µm band as done on Earth. KEYWORDS: Io, volcanic gases, silicon, fluorine, silicon tetrafluoride, condensates, geochemistry, atmospheric chemistry INTRODUCTION During our previous work on alkali halide chemistry on Io (Schaefer and Fegley, 2005b), we learned that silicon tetrafluoride is observed at several terrestrial volcanoes including Mount Iwodake, Vulcano, Mount Etna, and Popocatépetl (Francis et al., 1996; Love et al., 1998; Mori et al., 2002). Gaseous SiF4 is detected on Earth by observing its 9.7 µm band with infrared (IR) absorption spectroscopy, which is used to measure SiF4/SO2 molar ratios. -

(12) United States Patent (10) Patent No.: US 7,030,260 B2 Asirvatham Et Al
USOO7030260B2 (12) United States Patent (10) Patent No.: US 7,030,260 B2 Asirvatham et al. (45) Date of Patent: Apr. 18, 2006 (54) PREPARATION OF MIXED-HALOGEN Uhlig, W., Synthesis of Functional Substituted Oligosilanes HALO-SILANES Based on Sillyltriflate Derivatives, Organosilicon Chemistry: From Molecules to Materials, Eds. N. Auner, J. Weiss (75) Inventors: Edward Asirvatham, Chatham, NJ Federal Republic of Germany, (1994), pp. 21-26. (US); Jeff Czarnecki, Branchburg, NJ Katzenbeisser, U. Si-Si-Coupling Constants of Bromo (US); Matthew H. Luly, Hamburg, NY and Iododislanes and -trisilanes XSi-H and XSi-Hs. (US); Lawrence F. Mullan, (X= Br, I), Organosilicon Chemistry: From Molecules to Williamsville, NY (US); Alagappan Materials, Eds. N. Auner, J. Weiss, Federal Republic of Then appan, Wilmington, DE (US) Germany, (1994), pp. 37-38. (73) Assignee: Honeywell International Inc., Pátzold, U. Synthesis of Heavily Halogenated Vinylsilanes, Morristown, NJ (US) Organosilicon Chemistry IV: From Molecules to Materials, Eds. N. Auner, J. Weiss, Federal Republic of Germany, *) Notice: Subject to anyy disclaimer, the term of this (1996), pp. 226-228. patent is extended or adjusted under 35 Hassler, K., Köll, W., Synthesis and Spectroscopy of U.S.C. 154(b) by 214 days. Phenylated and Halogenated Trisilanes and Disilanes, Organosilicon Chemistry II: From Molecules to Materials, (21) Appl. No.: 10/377,367 Eds. N. Auner, J. Weiss, Federal Republic of Germany, (1996), pp. 81-88. (22) Filed: Feb. 27, 2003 Hassler, K. Schenzel, K., Syntheses, Si NMR Spectra, and (65) Prior Publication Data Vibrational Spectra of Methylated Trisilanes, Organosilicon Chemistry II: From Molecules to Materials, Eds. N. -

5.157 TABLE 5.29 Van Der Waals' Constants for Gases the Van Der
DEAN #37261 (McGHP) RIGHT INTERACTIVE top of rh PHYSICAL PROPERTIES 5.157 base of rh cap height TABLE 5.29 Van der Waals’ Constants for Gases base of text The van der Waals’ equation of state for a real gas is: na2 ͩͪP ϩ (V Ϫ nb) ϭ nRT for n moles V2 where P is the pressure, V the volume (in liters per mole ϭ 0.001 m3 per mole in the SI system), T the temperature (in degrees Kelvin), n the amount of substance (in moles), and R the gas constant. To use the values of a and b in the table, P must be expressed in the same units as in the gas constant. Thus, the pressure of a standard atmosphere may be expressed in the SI system as follows: 1 atm ϭ 101,325 N · mϪ2 ϭ 101,325 Pa ϭ 1.01325 bar The appropriate value for the gas constant is: 0.083 144 1 L · bar · KϪ1 · molϪ1 or 0.082 056 L · atm · KϪ1 · molϪ1 The van der Waals’ constants are related to the critical temperature and pressure, tc and Pc, in Table 6.5 by: 27 RT22 RT a ϭ ccand b ϭ 64 Pcc8 P Substance a,L2 · bar · molϪ2 b,L·molϪ1 Acetaldehyde 11.37 0.08695 Acetic acid 17.71 0.1065 Acetic anhydride 26.8 0.157 Acetone 16.02 0.1124 Acetonitrile 17.89 0.1169 Acetyl chloride 12.80 0.08979 Acetylene 4.516 0.05218 Acrylic acid 19.45 0.1127 Acrylonitrile 18.37 0.1222 Allene 8.235 0.07467 Allyl alcohol 15.17 0.1036 Aluminum trichloride 42.63 0.2450 2-Aminoethanol 7.616 0.0431 Ammonia 4.225 0.03713 Ammonium chloride 2.380 0.00734 Aniline 29.14 0.1486 Antimony tribromide 42.08 0.1658 Argon 1.355 0.03201 Arsenic trichloride 17.23 0.1039 Arsine 6.327 0.06048 Benzaldehyde 30.30 0.1553 Benzene 18.82 -

Silicon Tetrafluoride Interim AEGL Document
Silicon Tetrafluoride INTERIM: 06-2008 1 2 3 4 ACUTE EXPOSURE GUIDELINE LEVELS (AEGLs) 5 FOR 6 Silicon Tetrafluoride 7 (CAS Reg. No. 7783-61-1) 8 9 Si-F4 10 11 12 13 14 INTERIM 15 16 17 18 19 20 21 22 Silicon Tetrafluoride INTERIM: 06-2008/ Page 2 of 26 1 2 ACUTE EXPOSURE GUIDELINE LEVELS (AEGLs) 3 FOR 4 SILICON TETRAFLUORIDE 5 (CAS Reg. No. 7783-61-1) 6 7 8 INTERIM 9 10 Silicon Tetrafluoride INTERIM: 06-2008/ Page 3 of 26 1 2 PREFACE 3 4 Under the authority of the Federal Advisory Committee Act (FACA) P. L. 92-463 of 5 1972, the National Advisory Committee for Acute Exposure Guideline Levels for Hazardous 6 Substances (NAC/AEGL Committee) has been established to identify, review and interpret 7 relevant toxicologic and other scientific data and develop AEGLs for high priority, acutely toxic 8 chemicals. 9 10 AEGLs represent threshold exposure limits for the general public and are applicable to 11 emergency exposure periods ranging from 10 minutes to 8 hours. Three levels C AEGL-1, 12 AEGL-2 and AEGL-3 C are developed for each of five exposure periods (10 and 30 minutes, 1 13 hour, 4 hours, and 8 hours) and are distinguished by varying degrees of severity of toxic effects. 14 The three AEGLs are defined as follows: 15 16 AEGL-1 is the airborne concentration (expressed as parts per million or milligrams per 17 cubic meter [ppm or mg/m3]) of a substance above which it is predicted that the general 18 population, including susceptible individuals, could experience notable discomfort, irritation, or 19 certain asymptomatic, non-sensory effects. -
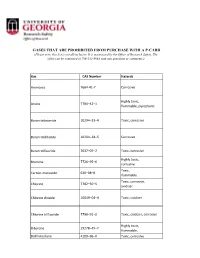
Lists of Gases for Pcard Manual.Xlsx
GASES THAT ARE PROHIBITED FROM PURCHASE WITH A P-CARD (Please note, this list is not all-inclusive. It is maintained by the Office of Research Safety. The office can be contacted at 706-542-9088 with any questions or comments.) Gas CAS Number Hazards Ammonia 7664‐41‐7 Corrosive Highly toxic, Arsine 7784–42–1 flammable, pyrophoric Boron tribromide 10294–33–4 Toxic, corrosive Boron trichloride 10294–34–5 Corrosive Boron trifluoride 7637–07–2 Toxic, corrosive Highly toxic, Bromine 7726–95–6 corrosive Toxic, Carbon monoxide 630–08–0 flammable Toxic, corrosive, Chlorine 7782–50–5 oxidizer Chlorine dioxide 10049–04–4 Toxic, oxidizer Chlorine trifluoride 7790–91–2 Toxic, oxidizer, corrosive Highly toxic, Diborane 19278–45–7 flammable, Dichlorosilane 4109–96–0 Toxic, corrosive Ethylene oxide 75–21–8 Toxic, flammable Highly toxic, corrosive, Fluorine 7782–41–4 oxidizer Highly toxic, Germane 7782–65–2 flammable Hydrogen bromide 10035–10–6 Toxic, corrosive Hydrogen chloride 7647–01–0 Toxic, corrosive Hydrogen cyanide 74–90–8 Highly toxic, flammable Hydrogen fluoride 7664–39–3 Toxic, corrosive Hydrogen iodide 10034‐85‐2 Toxic, corrosive Highly toxic, Hydrogen selenide 7783–07–5 flammable Toxic, flammable, Hydrogen sulfide 7783–06–4 corrosive Methyl bromide 74–83–9 Toxic Methyl isocyanate 624‐83‐9 Highly toxic, flammable Methyl mercaptan 74–93–1 Toxic, flammable Nickel carbonyl 13463–39–3 Highly toxic, flammable Highly toxic, Nitric oxide 10102–43–9 oxidizer Highly toxic, Nitrogen dioxide 10102–44–0 oxidizer, corrosive Highly toxic, Ozone 10028‐15‐6 -

Appendix a to Part 27
Appendix A to Part 27. – DHS Chemicals of Interest1 - - - WME - - – – Chemical Chemicals of Interest Abstract Synonym (COI) Service (CAS) # y Issue: Theft Theft y Issue: Screening Threshold Threshold Screening EXP/IEDP Issue: Security Sabotage/Contamination Security Issue: Theft Theft Issue: Security CWI/CWP Theft Issue: Security Release: Minimum Minimum Release: (%) Concentration Screening Release: (in Quantities Threshold pounds) Theft: Minimum (%) Concentration Theft: pounds (in Quantities noted) otherwise unless Minimum Sabotage: (%) Concentration Screening Sabotage: Quantities Threshold Release Issue: Security Toxic Release Issue: Security Flammables Release Issue: Security Explosives Securit Acetaldehyde 75-07-0 1.00 10,000 X Acetone cyanohydrin, 75-86-5 ACG APA X stabilized Acetyl bromide 506-96-7 ACG APA X Acetyl chloride 75-36-5 ACG APA X Acetyl iodine 507-02-8 ACG APA X Acetylene [Ethyne] 74-86-2 1.00 10,000 X Acrolein [2-Propenal] or Acrylaldehyde 107-02-8 1.00 5,000 X Acrylonitrile [2-Propenenitrile] 107-13-1 1.00 10,000 X Acrylyl chloride [2-Propenoyl Chloride] 814-68-6 1.00 10,000 X Allyl alcohol [2-Propen-1-ol] 107-18-6 1.00 15,000 X Allylamine [2-Propen-1-amine] 107-11-9 1.00 10,000 X Allyltrichlorosilane, 107-37-9 ACG APA X stabilized Aluminum (powder) 7429-90-5 ACG 100 X Aluminum bromide, 7727-15-3 ACG APA X anhydrous Aluminum chloride, 7446-70-0 ACG APA X anhydrous Aluminum phosphide 20859-73-8 ACG APA X Ammonia (anhydrous) 7664-41-7 1.00 10,000 X 1 The acronyms used in this appendix have the following meaning: ACG -

Abstract-A Siliceous Sublimate, Collected from an Active Fumarole of Kuju Volcano, Was Analyzed for Major Constituents
Geochemical Journal. Vol. 2, pp. 1 to 9, 1968. Silicon content of fumarolic gases and the formation of a siliceous sublimate FUMIHIRO HONDA and YOSHIHIKO MIZUTANI Department of Earth Sciences, Faculty of Science, Nagoya University, Chikusa, Nagoya, Japan (Received October 10, 1967; in revised form February 26, 1968) Abstract-A siliceous sublimate, collected from an active fumarole of Kuju Volcano, was analyzed for major constituents. The water soluble fraction of the sublimate contains large quantities of halogen acids and silicon, and the insoluble fraction consists of hydrated silica with a trace amount of native sulfur. In order to as certain the presence of gaseous silicon compounds in fumarolic gases, condensate samples were collected from high temperature fumaroles of Kuja and Nasudake Volcanoes with a silica-free sampling device. The silicon and fluorine contents of fumarolic gases range from 1 to 52 mg Si/kg H20 and 1 to 320 mg F/kg 1120, res pectively. Silicon is transported probably as fluoride in fumarolic gases, and the sublimate is formed by hydrolysis of silicon fluoride when the gas temperature falls to the boiling point of water. INTRODUCTION Fluorosilicic compounds are believed to play an important role in the trans portation of silicon in volcanic gases. LOVERING (1957) studied -the alteration of volcanic ash by the reaction with halogen acid at Valley of Ten Thousand Smokes, Alaska, and suggested that loss or gain of silica on the altered ash possibly re flected the attack of hydrogen fluoride or the hydrolysis of silicon fluoride. NABOKO (1957) also demonstrated the effect of gaseous fluorine compounds on basaltic rocks at Klyuchevskoy Volcano, Kamchatka. -

Silicon Tetrafluoride NOAL 0108 Country : SE / Language : EN
Page : 1/11 Revised edition no : 3.0 SAFETY DATA SHEET Revision date : 2021-06-30 Supersedes : 2020-07-17 Silicon tetrafluoride NOAL_0108 Country : SE / Language : EN SECTION 1: Identification of the substance/mixture and of the company/undertaking 1.1. Product identifier Trade name : Silicon tetrafluoride, SIF4 SDS no : NOAL_0108 Chemical description : Silicon tetrafluoride CAS-No. : 7783-61-1 EC-No. : 232-015-5 EC Index-No. : --- Registration-No. : Registration deadline not expired. Chemical formula : SiF4 1.2. Relevant identified uses of the substance or mixture and uses advised against Relevant identified uses : Industrial and professional uses. Perform risk assessment prior to use. Test gas/Calibration gas. Laboratory use. Chemical reaction / Synthesis. Use for manufacture of electronic/photovoltaic components. Contact supplier for more information on uses. Uses advised against : Consumer use. 1.3. Details of the supplier of the safety data sheet Company identification AIR LIQUIDE GAS AB Lundavägen 151 21209 Malmö - SWEDEN T +46 40 38 10 00 [email protected] E-Mail address (competent person) : [email protected] 1.4. Emergency telephone number Emergency telephone number : 112 Availability (24 / 7) SECTION 2: Hazards identification 2.1. Classification of the substance or mixture Classification according to Regulation (EC) No. 1272/2008 [CLP] Physical hazards Gases under pressure : Liquefied gas H280 Health hazards Acute toxicity (inhalation:gas) Category 2 H330 Skin corrosion/irritation, Category 1A H314 Serious eye damage/eye irritation, Category 1 H318 2.2. Label elements Labelling according to Regulation (EC) No. 1272/2008 [CLP] Hazard pictograms (CLP) : GHS04 GHS05 GHS06 HG2020002 Signal word (CLP) : Danger Hazard statements (CLP) : H280 - Contains gas under pressure; may explode if heated. -
SILICON TETRAFLUORIDE Safety Data Sheet
Revision Date 17-Mar-2015 , Version 1 _________________________________________________________________________________ SILICON TETRAFLUORIDE Safety Data Sheet _________________________________________________________________________________ 1. IDENTIFICATION Product identifier Product Name SILICON TETRAFLUORIDE Other means of identification Safety data sheet number LIND-P109 UN/ID no. UN1859 Synonyms Tetrafluorosilane; Silane, tetrafluoro-; Perfluorosilane; Silicon Fluoride Recommended use of the chemical and restrictions on use Recommended Use Industrial and professional use. Uses advised against Consumer use Details of the supplier of the safety data sheet Linde Gas North America LLC - Linde Merchant Production Inc. - Linde LLC 575 Mountain Ave. Murray Hill, NJ 07974 Phone: 908-464-8100 www.lindeus.com Linde Gas Puerto Rico, Inc. Road 869, Km 1.8 Barrio Palmas, Catano, PR 00962 Phone: 787-641-7445 www.pr.lindegas.com Linde Canada Limited 5860 Chedworth Way Mississauga, Ontario L5R 0A2 Phone: 905-501-1700 www.lindecanada.com * May include subsidiaries or affiliate companies/divisions. For additional product information contact your local customer service. Emergency telephone number Company Phone Number 800-232-4726 (Linde National Operations Center, US) 905-501-0802 (Canada) CHEMTREC: 1-800-424-9300 (North America) +1-703-527-3887 (International) _____________________________________________________________________________________________ Page 1 / 10 LIND-P109 SILICON TETRAFLUORIDE Revision Date 17-Mar-2015 _____________________________________________________________________________________________