Kartikeya Tiwari
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Rolllist Btech DD Bs2020batch
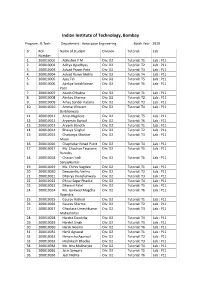
Indian Institute of Technology, Bombay Program : B.Tech. Department : Aerospace Engineering Batch Year : 2020 Sr. Roll Name of Student Division Tutorial Lab Number 1. 200010001 Abhishek P M Div: D2 Tutorial: T1 Lab : P11 2. 200010002 Aditya Upadhyay Div: D2 Tutorial: T2 Lab : P11 3. 200010003 Advait Pravin Pote Div: D2 Tutorial: T3 Lab : P11 4. 200010004 Advait Ranvir Mehla Div: D2 Tutorial: T4 Lab : P11 5. 200010005 Ajay Tak Div: D2 Tutorial: T5 Lab : P11 6. 200010006 Ajinkya Satishkumar Div: D2 Tutorial: T6 Lab : P11 Patil 7. 200010007 Akash Chhabra Div: D2 Tutorial: T1 Lab : P11 8. 200010008 Akshay Sharma Div: D2 Tutorial: T2 Lab : P11 9. 200010009 Amay Sunder Kataria Div: D2 Tutorial: T3 Lab : P11 10. 200010010 Ammar Khozem Div: D2 Tutorial: T4 Lab : P11 Barbhaiwala 11. 200010011 Anup Nagdeve Div: D2 Tutorial: T5 Lab : P11 12. 200010012 Aryaman Bansal Div: D2 Tutorial: T6 Lab : P11 13. 200010013 Aryank Banoth Div: D2 Tutorial: T1 Lab : P11 14. 200010014 Bhavya Singhal Div: D2 Tutorial: T2 Lab : P11 15. 200010015 Chaitanya Shankar Div: D2 Tutorial: T3 Lab : P11 Moon 16. 200010016 Chaphekar Ninad Punit Div: D2 Tutorial: T4 Lab : P11 17. 200010017 Ms. Chauhan Tejaswini Div: D2 Tutorial: T5 Lab : P11 Ramdas 18. 200010018 Chavan Yash Div: D2 Tutorial: T6 Lab : P11 Sanjaykumar 19. 200010019 Ms. Chinni Vagdevi Div: D2 Tutorial: T1 Lab : P11 20. 200010020 Deepanshu Verma Div: D2 Tutorial: T2 Lab : P11 21. 200010021 Dhairya Jhunjhunwala Div: D2 Tutorial: T3 Lab : P11 22. 200010022 Dhruv Sagar Phadke Div: D2 Tutorial: T4 Lab : P11 23. 200010023 Dhwanil Patel Div: D2 Tutorial: T5 Lab : P11 24. -

Some Questions Concerning the Ugc Course in Astrology
SOME QUESTIONS CONCERNING THE UGC COURSE IN ASTROLOGY Kushal Siddhanta 18 AUGUST 2001. Dr. Murali Manohar cient Indian astronomer Varahamihira who Joshi, the present Union HRD Minister and in the sixth century AD spoke about the a former professor of physics in the Alla- earth's rotation.” Citing another fact in sup- habad University, was invited to the IIT, port of introducing astrology as a university Kharagpur, as the chief guest at an open- course, he said, 16 universities of the coun- ing function organized on the occasion of try had already been offering astrology as its fiftieth foundation anniversary. The a subject in various forms. So his govern- West Bengal Chief Minister Mr. Buddhadeb ment is doing nothing new. People should Bhattacharya was another guest. not oppose the astrology and other courses only on political reasons. There may be de- As expected, Mr. Bhattacharya in his bates and arguments over the issue. The speech indirectly criticized the policy of the Government, he assured all, was ready to Union Government to introduce worn out hear. So on and so forth. subjects like astrology, paurohitya, vas- tushastra, yoga and human consciousness, It all had started when the UGC issued a etc., in the universities through the Univer- circular on 23 February 2001 with a pro- sity Grant Commission (UGC). Dr. Joshi posal to all the universities of the country in his address, however, did not go into to introduce UG and PG courses as well the trouble of explaining why his depart- as doctoral researches in Vedic Astrology ment was so keen on funding these anti- which it later renamed in parenthesis as quated subjects in the universities in spite Jyotirvigyan (astronomy). -
Sr.No Reference No AC No Part No Applicant Name 1 EJR602534753
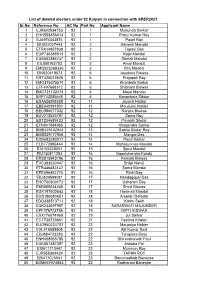
List of deleted electors under 92 Kalyani in connection with SRER2021 Sr.No Reference No AC No Part No Applicant Name 1 EJR602534753 92 1 Mukunda Sarkar 2 EHH558456014 92 1 Pratul Kumar Roy 3 EJA974343874 92 1 Payel Roy 4 EIU002027443 92 2 Ganesh Mandal 5 ETR614857838 92 2 Tapasi Das 6 EGP736388513 92 2 Kajal Mandal 7 EUB502386747 92 2 Shefali Mandal 8 EIL050163793 92 2 Amal Mandal 9 EMQ923268336 92 2 Rita Mondal 10 EIN820419573 92 6 Joyatsna Biswas 11 ERT425012526 92 6 Prajapati Roy 12 EMO375375574 92 6 Kiranbala Sarkar 13 EFF497668312 92 6 Shibnath Biswas 14 EMC721102474 92 6 Maya Mondal 15 EYF742085668 92 6 Kananbala Sikdar 16 ESA626393528 92 11 Jayanti Haldar 17 EBE640991801 92 11 Mousumi Halder 18 EEU986577332 92 12 Ranjita Biswas 19 EUV212535787 92 12 Soma Roy 20 EST234689433 92 12 Priyashi Sikdar 21 EFN841884965 92 12 Khagendra Sarkar 22 EME449402944 92 13 Sabita Sikdar Roy 23 EMS829177866 92 14 Mangal Das 24 ECN693282011 92 14 Parul Sarkar 25 ELB772896444 92 14 Maharunnisa Mandal 26 EAI105328251 92 15 Sipra Mandal 27 EIU150811283 92 16 Gopalchandra Kundu 28 ERS815943196 92 16 Kamala Biswas 29 EVC383532447 92 16 Shilpi Nandi 30 ETR446483147 92 16 Soma Mondal 31 EPE596482775 92 16 Rinki Bag 32 EBJ839599081 92 17 Nandagopal Das 33 EHC700803173 92 17 Usharani Dey 34 EKB685034245 92 17 Shiuli Biswas 35 EGK197602643 92 18 Harimati Mondal 36 EGS186680651 92 18 Ameran Dafadar 37 EDQ338513711 92 18 Karim Sekh 38 EUK233697997 92 18 SARASWATI MAJUMDER 39 EPF076723786 92 19 DIPTI BISWAS 40 EXX176074968 92 19 Jui Sarkar 41 ECT730729861 92 -

Mahabharata, Ramayana, Sita, Draupadi, Gandhari
Education 2014, 4(5): 122-125 DOI: 10.5923/j.edu.20140405.03 Sita (Character from the Indian epic –Ramayana), Draupadi and Gandhari (Characters from another Indian epic – Mahabharata) - A Comparative Study among Three Major Mythological Female Characters - Gandhari: An exception- Uditi Das1,*, Shamsad Begum Chowdhury1, Meejanur Rahman Miju2 1Institute of Education, Research and Training, University of Chittagong, Chittagong, Bangladesh 2Institute of Education, Research and Training (IERT), University of Chittagong, Chittagong, Bangladesh Abstract There are lots of female characters in Mahabharata and Ramayana but few characters enchant people of all ages and all classes. Mass people admit that Sita should be the icon of all women. Draupadi though a graceful character yet not to be imitated. Comparatively, Gandhari’s entrance into the epic is for a short while; though her appearance is very negligible, yet our research work is to show logically that Gandhari among these three characters is greater than the greatest. We think and have wanted to prove that Gandhari with her short appearance in the epic, excels all other female characters- depicted in Mahabharata and Ramayana. Keywords Mahabharata, Ramayana, Sita, Draupadi, Gandhari eighteen chapters. Again these chapters have been divided 1 . Introduction into one hundred sub-chapters. There are one lac (hundred thousand) verses in Mahabharata. Pandu, Kunti, Draupadi Ramayana: Ramayana is an epic composed by Valmiki and her five husbands, Dhritarastra, Gandhari and their one based on the life history of Ram-the king of the then Oudh hundred tyrannic sons – all are some of the famous and and is divided into seven cantos (Kanda). Sita was Ram’s notorious characters from this great epic. -

Undergraduate Syllabus
Presidency University Department of Philosophy Proposed Syllabus for UG Course in Philosophy Honours Sem. 1 PHIL 101: Paper Major-1 Western Logic 1 35+15 Marks PHIL 102: Paper Major-2 Indian Epistemology and Metaphysics 1 35+15 Marks Sem.2 PHIL 201: Paper Major-3, History of Western Epistemology and Metaphysics 35+15 Marks. PHIL 202: Paper Major-4 Western Logic 2 35+15 Marks Sem 3 PHIL 301: Paper Major-5, History of Western Epistemology and Metaphysics 2 35+15 Marks PHIL 302: Paper Major-6 Western Ethics 35+15 Marks PHIL 303: Paper Major-7, Indian Epistemology and Metaphysics 2 35+15 Marks Sem-4 PHIL 401: Paper Major-8 Philosophy of Language [Western] 35+15 Marks PHIL 402: Paper Major-9 Indian Logic 1 35+15 Marks PHIL 403: Paper Major-10 Philosophy of Mind 35+15 Marks Sem-5 PHIL 501: Paper Major-11 Epistemology & Metaphysics [western] 35+15 Marks PHIL 502: Paper Major-12 Verbal Knowledge- Indian Perspective 35+15 Marks PHIL 503: Paper Major-13 Western Logic 35+15 Marks PHIL 581: Paper Sessional 1 Presentation/seminar/work shop 50 Marks PHIL 582: Paper Sessional 2 Presentation/seminar/work shop 50 Marks Sem-6 PHIL 601: Paper Major-14 Philosophy of Religion 35+15 Marks PHIL 602: Paper Major-15 Applied Ethics 35+15 Marks PHIL 603: Paper Major-16 Western Logic 35+15 Marks PHIL 681: Paper Sessional 3 Presentation/seminar/work shop 50 Marks PHIL 682: Paper Sessional 4 Presentation/seminar/work shop 50 Marks 1 Sem. 1 PHIL 101: Paper Major-1 Western Logic 1 Marks 35+15 A. -

Handbook of Hinduism Ancient to Contemporary Books on the Related Theme by the Same Author
Handbook of Hinduism Ancient to Contemporary Books on the related theme by the Same Author ● Hinduism: A Gandhian Perspective (2nd Edition) ● Ethics for Our Times: Essays in Gandhian Perspective Handbook of Hinduism Ancient to Contemporary M.V. NADKARNI Ane Books Pvt. Ltd. New Delhi ♦ Chennai ♦ Mumbai Kolkata ♦ Thiruvananthapuram ♦ Pune ♦ Bengaluru Handbook of Hinduism: Ancient to Contemporary M.V. Nadkarni © Author, 2013 Published by Ane Books Pvt. Ltd. 4821, Parwana Bhawan, 1st Floor, 24 Ansari Road, Darya Ganj, New Delhi - 110 002 Tel.: +91(011) 23276843-44, Fax: +91(011) 23276863 e-mail: [email protected], Website: www.anebooks.com Branches Avantika Niwas, 1st Floor, 19 Doraiswamy Road, T. Nagar, Chennai - 600 017, Tel.: +91(044) 28141554, 28141209 e-mail: [email protected], [email protected] Gold Cornet, 1st Floor, 90 Mody Street, Chana Lane, (Mohd. Shakoor Marg), Opp. Masjid, Fort Mumbai - 400 001, Tel.: +91(022) 22622440, 22622441 e-mail: [email protected], [email protected] Flat No. 16A, 220 Vivekananda Road, Maniktala, Kolkata - 700 006, Tel.: +91(033) 23547119, 23523639 e-mail: [email protected] # 6, TC 25/2710, Kohinoor Flats, Lukes Lane, Ambujavilasam Road, Thiruvananthapuram - 01, Kerala, Tel.: +91(0471) 4068777, 4068333 e-mail: [email protected] Resident Representative No. 43, 8th ‘‘A’’ Cross, Ittumadhu, Banashankari 3rd Stage Bengaluru - 560 085, Tel.: +91 9739933889 e-mail: [email protected] 687, Narayan Peth, Appa Balwant Chowk Pune - 411 030, Mobile: 08623099279 e-mail: [email protected] Please be informed that the author and the publisher have put in their best efforts in producing this book. Every care has been taken to ensure the accuracy of the contents. -

How Buddhism Began by R.F. Gombrich
HOW BUDDHISM BEGAN This book, the second edition of How Buddhism Began, takes a fresh look at the earliest Buddhist texts and offers various suggestions how the teachings in them had developed. Two themes predominate. Firstly, it argues that we cannot understand the Buddha unless we understand that he was debating with other religious teachers, notably Brahmins. The other main theme concerns metaphor, allegory and literalism. By taking the words of the texts literally – despite the Buddha’s warning not to – successive generations of his disciples created distinctions and developed doctrines far beyond his original intention. This accessible, well-written book by one of the world’s top scholars in the field of Pali Buddhism is mandatory reading for all serious students of Buddhism. Richard F. Gombrich is Academic Director of the Oxford Centre for Buddhist Studies, and one of the most renowned Buddhist scholars in the world. From 1976 to 2004 he was Boden Professor of Sanskrit, University of Oxford. He has written extensively on Buddhism, including How Buddhism Began: The Conditioned Genesis of the Early Teachings (1996); Theravada Buddhism: A social history from ancient Benares to modern Colombo (1988); and with Gananath Obeyesekere, Buddhism transformed: Religious change in Sri Lanka (1988). He has been President of the Pali Text Society and was awarded the Sri Lanka Ranjana decoration by the President of Sri Lanka in 1994 and the SC Chakraborty medal by the Asiatic Society of Calcutta the previous year. Routledge Critical Studies in Buddhism General Editors: Charles S. Prebish and Damien Keown Routledge Critical Studies in Buddhism is a comprehensive study of the Buddhist tradition. -

The Impact Assessment of Diwali Fireworks Emissions on the Air Quality of a Tropical Urban Site, Hyderabad, India, During Three Consecutive Years
Environ Monit Assess DOI 10.1007/s10661-013-3102-x The impact assessment of Diwali fireworks emissions on the air quality of a tropical urban site, Hyderabad, India, during three consecutive years Venkata Swamy Yerramsetti & Anu Rani Sharma & Nikhil Gauravarapu Navlur & Venkanna Rapolu & N. S. K. Chitanya Dhulipala & P. R. Sinha Received: 15 June 2012 /Accepted: 16 January 2013 # Springer Science+Business Media Dordrecht 2013 Abstract Diwali is one of the largest festivals for attributed to firecrackers burning. The high correlation Hindu religion which falls in the period October– coefficient (~0.74) between NOx and SO2 concentra- November every year. During the festival days, exten- tions and higher SO2/NOx (S/N) index suggested air sive burning of firecrackers takes place, especially in quality degradation due to firecrackers burning. the evening hours, constituting a significant source of Furthermore, the Cloud–Aerosol Lidar and Infrared aerosols, black carbon (BC), organics, and trace gases. Pathfinder Satellite Observation-derived aerosol sub- The widespread use of sparklers was found to be typing map also confirmed the presence of smoke associated with short-term air quality degradation aerosols emitted from firecrackers burning over the events. The present study focuses on the influence of region. Nevertheless, the concentration level of pollu- Diwali fireworks emissions on surface ozone (O3), tants exhibited substantial decline over the region during nitrogen oxides (NOx), and BC aerosol concentration the years 2010 and 2011 compared to 2009 ascribed to over the tropical urban region of Hyderabad, India various awareness campaigns and increased cost of during three consecutive years (2009–2011). The trace firecrackers. -
Enrollment ID Student Name CHM out of 104 MTH out of 104 PHY out Of
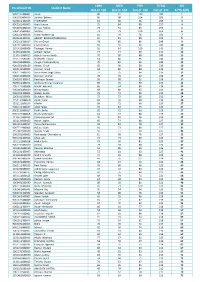
CHM MTH PHY TOTAL AIR Enrollment ID Student Name Out of 104 Out of 104 Out of 104 Out of 312 Ai²TS-1(XI) 9062191480001 Ayush 86 97 100 283 1 9062731480019 Sarthak Behera 80 98 104 282 2 9020611480035 P GIRINATH 83 80 96 259 3 9062161480005 Arpit Saxena 75 94 88 257 4 9062291480019 Shreya Pathak 78 91 85 254 5 9062191480002 Sarthak 73 79 100 252 6 9062161480036 Vishav Rakesh Vig 81 94 76 251 7 9020631480011 ABHIJIT BALASUBRAMANIAN 81 81 88 250 8 1062161580017 Pranav Goyal 78 85 86 249 9 9061811480069 Pratik Pranav 83 66 96 245 10 1151421680006 Tathagat Verma 59 84 100 243 11 1110551680161 Arihant Samar 77 74 90 241 12 1110161680015 Adarsh Kumar Banth 70 90 80 240 13 1152711680082 Siddharth Tiwary 64 80 96 240 13 9061931480004 Anagh Chattopadhyay 70 82 88 240 13 9061551480120 Adway Girish 69 90 80 239 16 1062121580009 Harman Singh 68 80 88 236 17 9062111480023 Simar Preet Singh Saluja 77 75 84 236 17 9062131480048 Navneel Singhal 78 76 82 236 17 9062201480041 Kartikeya Saxena 56 88 92 236 17 9020611480051 Andrews George Varghese 90 55 90 235 21 1061271580008 Hardik Agrawal 86 64 84 234 22 9061461480010 Nimay Gupta 68 80 85 233 23 9061101480005 Shalab Gupta 73 71 88 232 24 1062101580001 Shubham Mittal 72 72 88 232 24 1152131680070 Ayush Garg 84 56 90 230 26 1152111680155 Maulik 89 72 68 229 27 9061111480017 Ankit Yadav 70 84 75 229 27 9062131480017 Pulkit Sinha 65 72 92 229 27 9061471480058 Nischay Manwani 64 77 88 229 27 1110611680033 Praveenkumar M 70 81 78 229 27 1152121680105 Harish Jaglan 65 74 88 227 32 9061811480157 Tanay Raghavendra -

Ramayana and Mahabharata Deconstruction Literature Studies in Indonesia
Ramayana and Mahabharata Deconstruction Literature Studies in Indonesia Kundharu Saddhono, Budhi Setiawan and Kartika Rahmat Sari Dewi Graduate of Universitas Sebelas Maret, Surakarta, Indonesia [email protected] Keywords: Ramayana, Mahabharata, Deconstruction’s perspective, puppet, Sociology of Deconstruction Literature. Abstract: This study aims to describe Ramayana’s and Mahabarata’s Epos in deconstruction’s perspective with sociology of literature studies. This research is a descriptive qualitative study with of sociology of literature approach. The data source is some novel about puppet stories of Pitoyo Amrih. It employed the qualitative research design. Based on the analysis, the conclusion of the research are; (1) Antagonist characters of those Epic, had some kind in themself that can be best figure for the audience of the puppet’s show, (2) There’s lot of wisdom value as result of deconstruction’s analysis of the Epic, and (3) The assessment results of this study showed different sources of conflict, namely the seizure of the woman and the seizure of the kingdom. 1 INTRODUCTION diversity of Indonesia culture. S. Majumdar (2005: 182) also provides a similar case, concerning The term of multicultural is an appropriate calling priceless local wisdom legacy. name for Indonesia because its tradition, ethnicity, Javanese literary development is no longer language, art and other cultures diversity. The confined to the classical texts of previous poets, but diversity of culture apparently adds the value of has been widely transformed into various forms of culture richness of Indonesia to worldwide. Art is one modern literary works which still carries of treasure of the most famous cultural richness in Indonesia. -

Sangit-Bhavana, Visva Bharati Department of Hindusthani Classical Music
Sangit-Bhavana, Visva Bharati Department of Hindusthani Classical Music CURRICULUM FOR UNDERGRADUATE COURSE CHOICE BASED CREDIT SYSTEM S.No. COURSE SEMESTER CREDITS MARKS FULL MARKS Core Course 1 14 Courses I –VI 14X6=84 14X75 1050 08 Courses Practical 06 Courses Theoretical Discipline Specific Elective Course (DSE) 2 04 Courses V- VI 4X6=24 4X75 300 03Courses Practical 01 course Theoretical Generic Elective Course (GEC) 04 Courses I-IV 4X6=24 4X75 300 3 03 Courses Practical 01 Course Theoretical Skill Enhancement Compulsory Course (SECC) 4 III-IV 2X2=4 2X25 50 02 Courses Theoretical Ability Enhancement I-II 2X2=4 2X25 50 Compulsory Course (AECC) 5 02 Courses Theoretical 6 Foundation Course I-II 2X4=8 2X50 100 (Tagore Studies) 02 Courses Theoretical Total: 26+2=28 Courses - - - 1850 1 CHOICE BASED CREDIT SYSTEM B.MUS (HONS) IN HINDUSTHANI CLASSICAL MUSIC (VOCAL) COURSE AND MARKS DISTRIBUTION STRUCTURE Core course AECC SECC DSE GEC TS SEM Total Prac Theo Prac Theo Prac Theo Prac Theo Prac Theo Theo I 75 75 - 25 - - - - 75 - 50 300 II 75 75 - 25 - - - - 75 - 50 300 III 150 75 - - - 25 - - 75 - - 325 IV 150 75 - - - 25 - - - 75 - 325 V 75 75 - - - - 150 - - - - 300 VI 75 75 - - - - 75 75 - - - 300 Total 600 450 - 50 - 50 225 75 225 75 100 1850 2 Sangit-Bhavana, Visva Bharati Department of Hindusthani Classical Music CURRICULUM FOR UNDERGRADUATE COURSE CHOICE BASED CREDIT SYSTEM B.MUS (HONS) IN HINDUSTHANI CLASSICAL MUSIC TABLE OF CONTENTS HINDUSTHANI CLASSICAL MUSIC (VOCAL) 4 HINDUSTHANI CLASSICAL MUSIC INSTRUMENTAL (SITAR) 21 HINDUSTHANI -
SL No. Name of the Candidate Designation Organisation TNC No
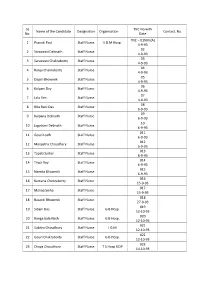
SL TNC No with Name of the Candidate Designation Organisation Contact. No No. Date TNC – 01NM (A) 1 Pranati Paul Staff Nurse S.D.M Hosp. 4-9-93 02 2 Saraswati Debnath Staff Nurse 4-9-93 03 3 Saraswati Chakraborty Staff Nurse 4-9-93 04 4 Ranju Chakraborty Staff Nurse 4-9-93 05 5 Dapili Bhowmik Staff Nurse 4-9-93 06 6 Kalyani Dey Staff Nurse 4-9-93 07 7 Lalu Sen Staff Nurse 4-9-93 08 8 Rita Rani Das Staff Nurse 6-9-93 09 9 Kalpana Debnath Staff Nurse 6-9-93 10 10 Jagatrani Debnath Staff Nurse 6-9-93 011 11 Gouri Lodh Staff Nurse 6-9-93 012 12 Manjistha Choudhary Staff Nurse 6-9-93 013 13 Tapati Sarkar Staff Nurse 6-9-93 014 14 Tripti Roy Staff Nurse 6-9-93 015 15 Namita Bhowmik Staff Nurse 6-9-93 016 16 Kamana Chokraborty Staff Nurse 15-9-93 017 17 Malina Sinha Staff Nurse 15-9-93 018 18 Basanti Bhowmik Staff Nurse 27-9-93 019 19 Sibani Das Staff Nurse G B Hosp. 12-10-93 020 20 Ranga bala Nath Staff Nurse G B Hosp. 12-10-93 021 21 Subhra Choudhury Staff Nurse I G M 12-10-93 022 22 Gouri Chakraborty Staff Nurse G B Hosp. 12-10-93 023 23 Chaya Choudhury Staff Nurse T S Hosp UDP 14-10-93 024 24 Sefali Patary Staff Nurse I G M 14-10-93 025 25 Amita Chakraborty Staff Nurse G B Hosp.