2 Toxicogenomics: Gene Expression Analysis and Computational Tools
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

UBQLN2 Polyclonal Antibody C-Terminal Ubiquitin-Associated Domain
UBQLN2 polyclonal antibody C-terminal ubiquitin-associated domain. They physically associate with both proteasomes and ubiquitin ligases, Catalog Number: PAB1699 and thus thought to functionally link the ubiquitination machinery to the proteasome to affect in vivo protein Regulatory Status: For research use only (RUO) degradation. This ubiquilin has also been shown to bind the ATPase domain of the Hsp70-like Stch protein. Product Description: Rabbit polyclonal antibody raised [provided by RefSeq] against synthetic peptide of UBQLN2. References: Immunogen: A synthetic peptide (conjugated with KLH) 1. Structural studies of the interaction between ubiquitin corresponding to C-terminus of human UBQLN2. family proteins and proteasome subunit S5a. Walters KJ, Kleijnen MF, Goh AM, Wagner G, Howley PM. Host: Rabbit Biochemistry. 2002 Feb 12;41(6):1767-77. 2. The hPLIC proteins may provide a link between the Reactivity: Human ubiquitination machinery and the proteasome. Kleijnen Applications: WB-Ce MF, Shih AH, Zhou P, Kumar S, Soccio RE, Kedersha (See our web site product page for detailed applications NL, Gill G, Howley PM. Mol Cell. 2000 Aug;6(2):409-19. information) 3. Selection system for genes encoding nuclear-targeted proteins. Ueki N, Oda T, Kondo M, Yano K, Noguchi T, Protocols: See our web site at Muramatsu M. Nat Biotechnol. 1998 http://www.abnova.com/support/protocols.asp or product Dec;16(13):1338-42. page for detailed protocols Form: Liquid Purification: Protein G purification Recommend Usage: Western Blot (1:1000) The optimal working dilution should be determined by the end user. Storage Buffer: In PBS (0.09% sodium azide) Storage Instruction: Store at 4°C. -

(Stch) Gene Reduces Prion Disease Incubation Time in Mice
Overexpression of the Hspa13 (Stch) gene reduces prion disease incubation time in mice Julia Grizenkovaa,b, Shaheen Akhtara,b, Holger Hummericha,b, Andrew Tomlinsona,b, Emmanuel A. Asantea,b, Adam Wenborna,b, Jérémie Fizeta,b, Mark Poultera,b, Frances K. Wisemanb, Elizabeth M. C. Fisherb, Victor L. J. Tybulewiczc, Sebastian Brandnerb, John Collingea,b, and Sarah E. Lloyda,b,1 aMedical Research Council (MRC) Prion Unit and bDepartment of Neurodegenerative Disease, University College London (UCL) Institute of Neurology, London WC1N 3BG, United Kingdom; and cDivision of Immune Cell Biology, MRC National Institute for Medical Research, London NW7 1AA, United Kingdom Edited by Reed B. Wickner, National Institutes of Health, Bethesda, MD, and approved July 10, 2012 (received for review May 28, 2012) Prion diseases are fatal neurodegenerative disorders that include way crosses and a heterogeneous cross (12–14). In these studies, bovine spongiform encephalopathy (BSE) and scrapie in animals the underlying functional polymorphism is unknown but could be and Creutzfeldt-Jakob disease (CJD) in humans. They are character- an amino acid change within the coding region of a protein or ized by long incubation periods, variation in which is determined by splicing variant or may occur within noncoding sequences such as many factors including genetic background. In some cases it is pos- untranslated regions, promoters, or other regulatory regions sible that incubation time may be directly correlated to the level of thereby influencing the pattern or level of expression. Although gene expression. To test this hypothesis, we combined incubation incubation time is a polygenic trait, it is possible that in some cases, time data from five different inbred lines of mice with quantitative there may be a direct correlation between incubation time and gene expression profiling in normal brains and identified five genes individual gene expression level. -

Development and Validation of a Serum Biomarker Panel for The
Journal of Cancer 2017, Vol. 8 2346 Ivyspring International Publisher Journal of Cancer 2017; 8(12): 2346-2355. doi: 10.7150/jca.19465 Research Paper Development and Validation of a Serum Biomarker Panel for the Detection of Esophageal Squamous Cell Carcinoma through RNA Transcriptome Sequencing Shan Xing1,2†, Xin Zheng1,2†, Li-Qiang Wei4†, Shi-Jian Song5, Dan Liu1,3, Ning Xue1,3, Xiao-Min Liu1,2, Mian-Tao Wu1,2, Qian Zhong1,3, Chu-Mei Huang6, Mu-Sheng Zeng1,3, Wan-Li Liu1,2 1. State Key Laboratory of Oncology in Southern China, Collaborative Innovation Center for Cancer Medicine, Guangzhou, China 2. Department of Clinical Laboratory, Sun Yat-sen University Cancer Center, Guangzhou, China 3. Department of Experimental Research, Sun Yat-sen University cancer center, Guangzhou, China 4. Department of Clinical Laboratory, Shaanxi Provincial People’s Hospital, Xian, China 5. Guangdong Experimental High School, Guangzhou, China 6. Department of Laboratory Medicine, Sun Yat-sen University First Affiliated Hospital, Guangzhou, China † Xing S, Zheng X and Wei LQ contributed equally to this work Corresponding authors: Wan-Li Liu. Email: [email protected]; Phone: 86-020-87343196. Department of Clinical Laboratory, Sun Yat-sen University Cancer Center, 651 Dongfeng Road East,Guangzhou 510060, P.R. China. Mu-Sheng Zeng; Email: [email protected] Phone: +86-020-87343192. Department of Experimental Research, Sun Yat-sen University cancer center, 651 Dongfeng Road East,Guangzhou 510060, P.R.China © Ivyspring International Publisher. This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). -

RTL8 Promotes Nuclear Localization of UBQLN2 to Subnuclear Compartments Associated with Protein Quality Control
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.21.440788; this version posted April 22, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: RTL8 promotes nuclear localization of UBQLN2 to subnuclear compartments associated with protein quality control Harihar Milaganur Mohan1,2, Amit Pithadia1, Hanna Trzeciakiewicz1, Emily V. Crowley1, Regina Pacitto1 Nathaniel Safren1,3, Chengxin Zhang4, Xiaogen Zhou4, Yang Zhang4, Venkatesha Basrur5, Henry L. Paulson1,*, Lisa M. Sharkey1,* 1. Department of Neurology, University of Michigan, Ann Arbor, MI 48109-2200 2. Graduate Program in Cellular and Molecular Biology, University of Michigan, Ann Arbor, MI 48109- 2200 3. Present address: Department of Neurology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611 4. Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109-2200 5. Department of Pathology, University of Michigan Medical School, Ann Arbor, MI 48109. *Corresponding authors: Henry Paulson: Department of Neurology, University of Michigan, Ann Arbor, MI 48109-2200; [email protected]; Tel. (734) 615-5632; Fax. (734) 615-5655 Lisa M Sharkey: Department of Neurology, University of Michigan, Ann Arbor, MI 48109-2200; [email protected]; Tel. (734) 763-3496; Fax (734) 615-5655 Keywords: Ubiquilin, UBQLN2, RTL8, Nuclear Protein Quality Control, Ubiquitin Proteasome System Declarations Funding: This work was supported by NIH 9R01NS096785-06, 1P30AG053760-01, The Amyotrophic Lateral Sclerosis Foundation and the UM Protein Folding Disease Initiative. Conflicts of interest/Competing interests: None declared Availability of data and material: • The authors confirm that the data supporting the findings in this are available within the article, at repository links provided within the article, and within its supplementary files. -

Differential Effects of STCH and Stress-Inducible Hsp70 on the Stability and Maturation of NKCC2
International Journal of Molecular Sciences Article Differential Effects of STCH and Stress-Inducible Hsp70 on the Stability and Maturation of NKCC2 Dalal Bakhos-Douaihy 1,2, Elie Seaayfan 1,2,† , Sylvie Demaretz 1,2,†, Martin Komhoff 3,‡ and Kamel Laghmani 1,2,*,‡ 1 Centre de Recherche des Cordeliers, INSERM, Sorbonne Université, USPC, Université Paris Descartes, Université Paris Diderot, 75006 Paris, France; [email protected] (D.B.-D.); [email protected] (E.S.); [email protected] (S.D.) 2 CNRS, ERL8228, 75006 Paris, France 3 University Children’s Hospital, Philipps-University, 35043 Marburg, Germany; [email protected] * Correspondence: [email protected]; Tel.: +33-1-44-27-50-68; Fax: +33-1-44-27-51-19 † These authors contributed equally to this work. ‡ These authors contributed equally to this work. Abstract: Mutations in the Na-K-2Cl co-transporter NKCC2 lead to type I Bartter syndrome, a life- threatening kidney disease. We previously showed that export from the ER constitutes the limiting step in NKCC2 maturation and cell surface expression. Yet, the molecular mechanisms involved in this process remain obscure. Here, we report the identification of chaperone stress 70 protein (STCH) and the stress-inducible heat shock protein 70 (Hsp70), as two novel binding partners of the ER-resident form of NKCC2. STCH knock-down increased total NKCC2 expression whereas Hsp70 knock-down or its inhibition by YM-01 had the opposite effect. Accordingly, overexpressing of STCH and Hsp70 exerted opposite actions on total protein abundance of NKCC2 and its folding mutants. -
Download Validation Data
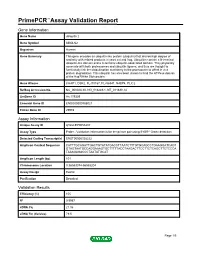
PrimePCR™Assay Validation Report Gene Information Gene Name ubiquilin 2 Gene Symbol UBQLN2 Organism Human Gene Summary This gene encodes an ubiquitin-like protein (ubiquilin) that shares high degree of similarity with related products in yeast rat and frog. Ubiquilins contain a N-terminal ubiquitin-like domain and a C-terminal ubiquitin-associated domain. They physically associate with both proteasomes and ubiquitin ligases; and thus are thought to functionally link the ubiquitination machinery to the proteasome to affect in vivo protein degradation. This ubiquilin has also been shown to bind the ATPase domain of the Hsp70-like Stch protein. Gene Aliases CHAP1, DSK2, FLJ10167, FLJ56541, N4BP4, PLIC2 RefSeq Accession No. NC_000023.10, NG_016249.1, NT_011630.14 UniGene ID Hs.179309 Ensembl Gene ID ENSG00000188021 Entrez Gene ID 29978 Assay Information Unique Assay ID qHsaCEP0055207 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000338222 Amplicon Context Sequence CATTTGCAGATTGACTGTATATGACCTTAATCTTTGTGCAGCCTGAAGGATCAGT GTAGTAATGCCAGGAAAGTGCTTTTTACCTAAGACTTCCTTCTCAGCTTCTCCCA TAAAGAGACCCTAATATGCAT Amplicon Length (bp) 101 Chromosome Location X:56593074-56593204 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 105 R2 0.9967 cDNA Cq 21.06 cDNA Tm (Celsius) 79.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 25.2 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay -

DNA Replication Is Altered in Immunodeficiency
European Journal of Human Genetics (2012) 20, 1044–1050 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE DNA replication is altered in Immunodeficiency Centromeric instability Facial anomalies (ICF) cells carrying DNMT3B mutations Erica Lana1,2, Andre´ Me´garbane´3,4,He´le`ne Tourrie`re5, Pierre Sarda6,Ge´rard Lefranc5,7, Mireille Claustres1,2,6 and Albertina De Sario*,1,2 ICF syndrome is a rare autosomal recessive disorder that is characterized by Immunodeficiency, Centromeric instability, and Facial anomalies. In all, 60% of ICF patients have mutations in the DNMT3B (DNA methyltransferase 3B) gene, encoding a de novo DNA methyltransferase. In ICF cells, constitutive heterochromatin is hypomethylated and decondensed, metaphase chromosomes undergo rearrangements (mainly involving juxtacentromeric regions), and more than 700 genes are aberrantly expressed. This work shows that DNA replication is also altered in ICF cells: (i) heterochromatic genes replicate earlier in the S-phase; (ii) global replication fork speed is higher; and (iii) S-phase is shorter. These replication defects may result from chromatin changes that modify DNA accessibility to the replication machinery and/or from changes in the expression level of genes involved in DNA replication. This work highlights the interest of using ICF cells as a model to investigate how DNA methylation regulates DNA replication in humans. European Journal of Human Genetics (2012) 20, 1044–1050. doi:10.1038/ejhg.2012.41; published online 29 February 2012 Keywords: ICF syndrome; DNA replication; DNA methylation INTRODUCTION sequences (ie, satellites 2 and 3, subtelomeric sequences, and Alu With the availability of emerging technologies in genomics, a major sequences)8–10 and genes located in constitutive and facultative hetero- challenge has become to understand how epigenetic modifications chromatin (hereafter named C-heterochromatin and F-heterochroma- regulate the genome function. -

Rabbit Anti-STCH/HSPA13/FITC Conjugated Antibody
SunLong Biotech Co.,LTD Tel: 0086-571- 56623320 Fax:0086-571- 56623318 E-mail:[email protected] www.sunlongbiotech.com Rabbit Anti-STCH/HSPA13/FITC Conjugated antibody SL12823R-FITC Product Name: Anti-STCH/HSPA13/FITC Chinese Name: FITC标记的热休克蛋白70家族13抗体 Heat shock 70 kDa protein 13; HEAT SHOCK 70-KD PROTEIN 13; heat shock protein 70kDa family, member 13; heat shock protein70kDa family, member13; HSP13_HUMAN; HSPA13; MGC133835; Microsomal stress 70 protein ATPase core; Alias: Microsomal stress-70 protein ATPase core; STCH; Stress 70 protein chaperone microsome associated 60 kDa protein; Stress-70 protein chaperone microsome- associated 60 kDa protein. Organism Species: Rabbit Clonality: Polyclonal React Species: Human,Mouse,Rat,Dog,Pig,Cow,Horse,Rabbit,Sheep,Chimpanzee, ICC=1:50-200IF=1:50-200 Applications: not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. Molecular weight: 50kDa Form: Lyophilizedwww.sunlongbiotech.com or Liquid Concentration: 2mg/1ml immunogen: KLH conjugated synthetic peptide derived from human STCH/HSPA13 Lsotype: IgG Purification: affinity purified by Protein A Storage Buffer: 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year Storage: when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C. background: Product Detail: The protein encoded by this gene is a member of the heat shock protein 70 family and is found associated with microsomes. -

Tetrasomy 21Pterrq21.2 in a Male Infant Without Typical Down's Syndrome Dysmorphic Features but Moderate Mental Retardation
1of6 ELECTRONIC LETTER J Med Genet: first published as 10.1136/jmg.2003.011833 on 1 March 2004. Downloaded from Tetrasomy 21pterRq21.2 in a male infant without typical Down’s syndrome dysmorphic features but moderate mental retardation I Rost, H Fiegler, C Fauth, P Carr, T Bettecken, J Kraus, C Meyer, A Enders, A Wirtz, T Meitinger, N P Carter, M R Speicher ............................................................................................................................... J Med Genet 2004;41:e26 (http://www.jmedgenet.com/cgi/content/full/41/3/e26). doi: 10.1136/jmg.2003.011833 own’s syndrome is caused by trisomy of chromosome 21. This invariably results in cognitive impairment, Key points Dhypotonia, and characteristic phenotypic features such as flat facies, upslanting palpebral fissures, and inner N Genetic investigation of patients with a partial chromo- epicanthal folds, and variations in digits and the ridge some 21 imbalance should help define which genes of formation on hands and feet. Furthermore, trisomy 21 is a chromosome 21 play a key role in the various risk factor for congenital heart disease, Hirschsprung’s symptoms of Down’s syndrome. disease, and many other developmental abnormalities.1 N We present a male infant with an additional marker The physical phenotype of Down’s syndrome has often chromosome derived from chromosome 21. The been attributed to an imbalance of the region comprising patient’s phenotype was not characteristic of Down’s bands in chromosome region q22.12q22.3.1 However, imbal- ance of other regions on chromosome 21 may also contribute syndrome; however, he had moderate mental retarda- to the phenotype. A ‘‘phenotypic map’’ established from cell tion, comparable to the cognitive defects usually seen lines from patients with partial trisomy 21 suggested that in Down’s syndrome patients. -

Anti-UBQLN2 Antibody
Anti-UBQLN2 Antibody Alternative Names: ALS15, CHAP1, DSK2, HRIHFB2157, N4BP4, PLIC2, Ubiquilin-2 Catalogue Number: AB18-10055-50ug Size: 50 µg Background Information Ubiquilin-2 (UBQLN2) is a 624-amino acid multi-domain adaptor protein and a member of the ubiquilin family of proteins that regulate the degradation of ubiquitinated proteins by the ubiquitin-proteasome system (UPS), autophagy and the endoplasmic reticulum- associated protein degradation (ERAD) pathway. Ubiquilins are characterised by the presence of an N-terminal ubiquitin-like domain and a C-terminal ubiquitin-associated domain. The central portion is highly variable. UBQLN2 Mediates the proteasomal targeting of misfolded or accumulated proteins for degradation by binding to their polyubiquitin chains, through the ubiquitin-associated domain (UBA) and by interacting with the subunits of the proteasome through the ubiquitin- like domain (ULD). Mutations in UBQLN2 are associated with Amyotrophic Lateral Sclerosis with most ALS-linked mutations localised to the proline-rich repeat (Pxx) region that is unique to ubiquilin-2 and not present in the other members of the ubiquilin protein family. UBQLN2 has also been shown to bind the ATPase domain of the Hsp70-like Stch protein. Mutations in UBQLN2 are also observed in familial ALS (FALS) cases associated with aberrant TDP-43 inclusions. Product Information Antibody Type: Polyclonal Host: Rabbit Isotype: IgG Species Reactivity: Human, Mouse Immunogen: Partial length recombinant human UBQLN2 from the N-terminal region Format: 50 µg in 50 µl PBS containing 0.02% sodium azide. Storage Conditions: 6 months: 4°C. Long-term storage: -20°C. Avoid multiple freeze and thaw cycles. Applications: WB WB 1:200-2000. -

Overexpression of the Hspa13 (Stch) Gene Reduces Prion Disease Incubation Time in Mice
Overexpression of the Hspa13 (Stch) gene reduces prion disease incubation time in mice Julia Grizenkovaa,b, Shaheen Akhtara,b, Holger Hummericha,b, Andrew Tomlinsona,b, Emmanuel A. Asantea,b, Adam Wenborna,b, Jérémie Fizeta,b, Mark Poultera,b, Frances K. Wisemanb, Elizabeth M. C. Fisherb, Victor L. J. Tybulewiczc, Sebastian Brandnerb, John Collingea,b, and Sarah E. Lloyda,b,1 aMedical Research Council (MRC) Prion Unit and bDepartment of Neurodegenerative Disease, University College London (UCL) Institute of Neurology, London WC1N 3BG, United Kingdom; and cDivision of Immune Cell Biology, MRC National Institute for Medical Research, London NW7 1AA, United Kingdom Edited by Reed B. Wickner, National Institutes of Health, Bethesda, MD, and approved July 10, 2012 (received for review May 28, 2012) Prion diseases are fatal neurodegenerative disorders that include way crosses and a heterogeneous cross (12–14). In these studies, bovine spongiform encephalopathy (BSE) and scrapie in animals the underlying functional polymorphism is unknown but could be and Creutzfeldt-Jakob disease (CJD) in humans. They are character- an amino acid change within the coding region of a protein or ized by long incubation periods, variation in which is determined by splicing variant or may occur within noncoding sequences such as many factors including genetic background. In some cases it is pos- untranslated regions, promoters, or other regulatory regions sible that incubation time may be directly correlated to the level of thereby influencing the pattern or level of expression. Although gene expression. To test this hypothesis, we combined incubation incubation time is a polygenic trait, it is possible that in some cases, time data from five different inbred lines of mice with quantitative there may be a direct correlation between incubation time and gene expression profiling in normal brains and identified five genes individual gene expression level. -

Ep 2327796 A1
(19) & (11) EP 2 327 796 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 01.06.2011 Bulletin 2011/22 C12Q 1/68 (2006.01) (21) Application number: 10184813.3 (22) Date of filing: 09.06.2004 (84) Designated Contracting States: • Spira, Avrum AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Newton, Massachusetts 02465 (US) HU IE IT LI LU MC NL PL PT RO SE SI SK TR (74) Representative: Brown, David Leslie (30) Priority: 10.06.2003 US 477218 P Haseltine Lake LLP Redcliff Quay (62) Document number(s) of the earlier application(s) in 120 Redcliff Street accordance with Art. 76 EPC: Bristol 04776438.6 / 1 633 892 BS1 6HU (GB) (71) Applicant: THE TRUSTEES OF BOSTON Remarks: UNIVERSITY •This application was filed on 30-09-2010 as a Boston, MA 02218 (US) divisional application to the application mentioned under INID code 62. (72) Inventors: •Claims filed after the date of filing of the application/ • Brody, Jerome S. after the date of receipt of the divisional application Newton, Massachusetts 02458 (US) (Rule 68(4) EPC). (54) Detection methods for disorders of the lung (57) The present invention is directed to prognostic vides a minimally invasive sample procurement method and diagnostic methods to assess lung disease risk in combination with the gene expression-based tools for caused by airway pollutants by analyzing expression of the diagnosis and prognosis of diseases of the lung, par- one or more genes belonging to the airway transcriptome ticularly diagnosis and prognosis of lung cancer provided herein.