Molecular and Genetic Mechanism of Non-Syndromic Congenital Cataracts
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Isyte: Integrated Systems Tool for Eye Gene Discovery
Lens iSyTE: Integrated Systems Tool for Eye Gene Discovery Salil A. Lachke,1,2,3,4 Joshua W. K. Ho,1,4,5 Gregory V. Kryukov,1,4,6 Daniel J. O’Connell,1 Anton Aboukhalil,1,7 Martha L. Bulyk,1,8,9 Peter J. Park,1,5,10 and Richard L. Maas1 PURPOSE. To facilitate the identification of genes associated ther investigation. Extension of this approach to other ocular with cataract and other ocular defects, the authors developed tissue components will facilitate eye disease gene discovery. and validated a computational tool termed iSyTE (integrated (Invest Ophthalmol Vis Sci. 2012;53:1617–1627) DOI: Systems Tool for Eye gene discovery; http://bioinformatics. 10.1167/iovs.11-8839 udel.edu/Research/iSyTE). iSyTE uses a mouse embryonic lens gene expression data set as a bioinformatics filter to select candidate genes from human or mouse genomic regions impli- ven with the advent of high-throughput sequencing, the cated in disease and to prioritize them for further mutational Ediscovery of genes associated with congenital birth defects and functional analyses. such as eye defects remains a challenge. We sought to develop METHODS. Microarray gene expression profiles were obtained a straightforward experimental approach that could facilitate for microdissected embryonic mouse lens at three key devel- the identification of candidate genes for developmental disor- opmental time points in the transition from the embryonic day ders, and, as proof-of-principle, we chose defects involving the (E)10.5 stage of lens placode invagination to E12.5 lens primary ocular lens. Opacification of the lens results in cataract, a leading cause of blindness that affects 77 million persons and fiber cell differentiation. -

A New Mutation in BFSP2 (G1091A) Causes Autosomal Dominant Congenital Lamellar Cataracts
Molecular Vision 2008; 14:1906-1911 <http://www.molvis.org/molvis/v14/a226> © 2008 Molecular Vision Received 17 August 2008 | Accepted 18 October 2008 | Published 24 October 2008 A new mutation in BFSP2 (G1091A) causes autosomal dominant congenital lamellar cataracts Xu Ma,1,2,3 Fei-Feng Li,1,2 Shu-Zhen Wang,4 Chang Gao,1,2 Meng Zhang,2 Si-Quan Zhu4 (The first three authors contributed equally to this work.) 1Graduate School, Peking Union Medical College, Beijing, China; 2Department of Genetics, National Research Institute for Family Planning, Beijing, China; 3WHO Collaborative Center for Research in Human Reproduction, Beijing, China; 4Beijing Tongren Eye Center, Capital Medical University, Beijing, China Purpose: We sought to identify the genetic defect in a four-generation Chinese family with autosomal dominant congenital lamellar cataracts and demonstrate the functional analysis with biosoftware of a candidate gene in the family. Methods: Family history data were recorded. Clinical and ophthalmologic examinations were performed on family members. All the members were genotyped with microsatellite markers at loci considered to be associated with cataracts. Two-point LOD scores were calculated by using the Linkage Software after genotyping. A mutation was detected by using gene-specific primers in direct sequencing. Wild type and mutant proteins were analyzed with Online Bio-Software. Results: Affected members of this family had lamellar cataracts. Linkage analysis was obtained at markers D3S2322 (LOD score [Z]=7.22, recombination fraction [θ]=0.0) and D3S1541 (Z=5.42, θ=0.0). Haplotype analysis indicated that the cataract gene was closely linked to these two markers. -

Post-Mortem Whole-Exome Analysis in a Large Sudden Infant Death Syndrome Cohort with a Focus on Cardiovascular and Metabolic Genetic Diseases
European Journal of Human Genetics (2017) 25, 404–409 & 2017 Macmillan Publishers Limited, part of Springer Nature. All rights reserved 1018-4813/17 www.nature.com/ejhg ARTICLE Post-mortem whole-exome analysis in a large sudden infant death syndrome cohort with a focus on cardiovascular and metabolic genetic diseases Jacqueline Neubauer*,1, Maria Rita Lecca2, Giancarlo Russo2, Christine Bartsch3, Argelia Medeiros-Domingo4, Wolfgang Berger5,6,7 and Cordula Haas1 Sudden infant death syndrome (SIDS) is described as the sudden and unexplained death of an apparently healthy infant younger than one year of age. Genetic studies indicate that up to 35% of SIDS cases might be explained by familial or genetic diseases such as cardiomyopathies, ion channelopathies or metabolic disorders that remained undetected during conventional forensic autopsy procedures. Post-mortem genetic testing by using massive parallel sequencing (MPS) approaches represents an efficient and rapid tool to further investigate unexplained death cases and might help to elucidate pathogenic genetic variants and mechanisms in cases without a conclusive cause of death. In this study, we performed whole-exome sequencing (WES) in 161 European SIDS infants with focus on 192 genes associated with cardiovascular and metabolic diseases. Potentially causative variants were detected in 20% of the SIDS cases. The majority of infants had variants with likely functional effects in genes associated with channelopathies (9%), followed by cardiomyopathies (7%) and metabolic diseases (1%). Although lethal arrhythmia represents the most plausible and likely cause of death, the majority of SIDS cases still remains elusive and might be explained by a multifactorial etiology, triggered by a combination of different genetic and environmental risk factors. -

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -
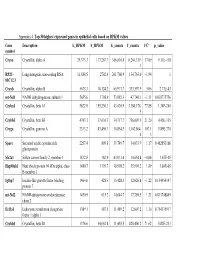
Appendix 4. Top 50 Highest Expressed Genes in Epithelial Cells Based on RPKM Values
Appendix 4. Top 50 highest expressed genes in epithelial cells based on RPKM values Gene Description E_RPKM F_RPKM E_counts F_counts FC* p_value symbol Cryaa Crystallin, alpha A 29,373.3 177,267.7 366,616.4 6,264,319. 17.09 9.11E-118 1 RP23– Long intergenic non-coding RNA 11,888.5 2702.4 261,760.9 134,763.0 −1.94 1 81C12.3 Cryab Crystallin, alpha B 5673.3 10,124.2 65,971.7 333,597.9 5.06 2.71E-43 mt-Nd1 NADH dehydrogenase, subunit 1 5655.6 1798.9 53,082.3 47,748.1 −1.11 0.838775756 Cryba1 Crystallin, beta A1 5622.0 155,230.3 43,420.9 3,380,176. 77.85 1.34E-240 5 Crybb3 Crystallin, beta B3 4743.1 37,636.3 34,717.7 736,007.9 21.20 4.45E-135 Cryga Crystallin, gamma A 2333.2 83,496.3 10,854.5 1,162,864. 107.1 5.89E-270 6 3 Sparc Secreted acidic cysteine rich 2257.4 809.8 39,749.7 34,033.9 −1.17 0.462853166 glycoprotein Slc2a1 Solute carrier family 2, member 1 1832.8 162.9 43,031.4 10,654.8 −4.04 1.67E-05 Hsp90ab1 Heat shock protein 90 kDa alpha, class 1480.7 1139.7 18,998.2 35,901.2 1.89 3.84E-05 B member 1 Igfbp7 Insulin-like growth factor binding 1464.6 428.3 15,428.3 12,626.8 −1.22 0.154954147 protein 7 mt-Nd2 NADH-ubiquinone oxidoreductase 1450.9 615.2 14,644.7 17,789.5 1.21 0.833748849 chain 2 Eef1a1 Eukaryotic translation elongation 1389.1 587.5 11,489.2 12,607.2 1.10 0.754135917 factor 1 alpha 1 Crybb1 Crystallin, beta B1 1376.6 34,662.8 11,455.5 820,406.2 71.62 5.82E-233 Htra3 HtrA serine peptidase 3 1338.6 162.0 23,197.6 6433.9 −3.61 3.93E-05 Gnb2l1 Guanine nucleotide-binding protein 1293.3 670.1 14,495.1 21,652.1 1.49 0.001685952 -

Variants in PAX6, PITX3 and HSF4 Causing Autosomal Dominant Congenital Cataracts ✉ ✉ Vanita Berry 1,2 , Alex Ionides2, Nikolas Pontikos 1,2, Anthony T
www.nature.com/eye ARTICLE OPEN Variants in PAX6, PITX3 and HSF4 causing autosomal dominant congenital cataracts ✉ ✉ Vanita Berry 1,2 , Alex Ionides2, Nikolas Pontikos 1,2, Anthony T. Moore2, Roy A. Quinlan3 and Michel Michaelides 1,2 © Crown 2021 BACKGROUND: Lens development is orchestrated by transcription factors. Disease-causing variants in transcription factors and their developmental target genes are associated with congenital cataracts and other eye anomalies. METHODS: Using whole exome sequencing, we identified disease-causing variants in two large British families and one isolated case with autosomal dominant congenital cataract. Bioinformatics analysis confirmed these disease-causing mutations as rare or novel variants, with a moderate to damaging pathogenicity score, with testing for segregation within the families using direct Sanger sequencing. RESULTS: Family A had a missense variant (c.184 G>A; p.V62M) in PAX6 and affected individuals presented with nuclear cataract. Family B had a frameshift variant (c.470–477dup; p.A160R*) in PITX3 that was also associated with nuclear cataract. A recurrent missense variant in HSF4 (c.341 T>C; p.L114P) was associated with congenital cataract in a single isolated case. CONCLUSIONS: We have therefore identified novel variants in PAX6 and PITX3 that cause autosomal dominant congenital cataract. Eye; https://doi.org/10.1038/s41433-021-01711-x INTRODUCTION consistent with early developmental effects as would be Cataract the opacification of the eye lens is the most common, but anticipated for PAX6 and PITX3 transcription factors. Recently, treatable cause of blindness in the world (https://www.who.int/ we have found two novel mutations in the transcription factors publications-detail/world-report-on-vision). -

Cardiomyopathy
UNIVERSITY OF MINNESOTA PHYSICIANS OUTREACH LABS Submit this form along with the appropriate MOLECULAR DIAGNOSTICS (612) 273-8445 Molecular requisition (Molecular Diagnostics or DATE: TIME COLLECTED: PCU/CLINIC: Molecular NGS Oncology). AM PM PATIENT IDENTIFICATION DIAGNOSIS (Dx) / DIAGNOSIS CODES (ICD-9) - OUTPATIENTS ONLY SPECIMEN TYPE: o Blood (1) (2) (3) (4) PLEASE COLLECT 5-10CC IN ACD-A OR EDTA TUBE ORDERING PHYSICIAN NAME AND PHONE NUMBER: Tests can be ordered as a full panel, or by individual gene(s). Please contact the genetic counselor with any questions at 612-624-8948 or by pager at 612-899-3291. _______________________________________________ Test Ordered- EPIC: Next generation sequencing(Next Gen) Sunquest: NGS Brugada syndrome DMD Aortopathy Full panel DNAJC19 SCN5A DSP Full panel GPD1L Cardiomyopathy, familial MYH11 CACNA1C hypertrophic ACTA2 SCN1B Full panel MYLK KCNE3 ANKRD1 FBN2 SCN3B JPH2 SLC2A10 HCN4 PRKAG2 COL5A2 MYH7 COL5A1 MYL2 COL3A1 Cardiomyopathy ACTC1 CBS Cardiomyopathy, dilated CSRP3 SMAD3 Full panel TNNC1 TGFBR1 LDB3 MYH6 TGFBR2 LMNA VCL FBN1 ACTN2 MYOZ2 Arrhythmogenic right ventricular DSG2 PLN dysplasia NEXN CALR3 TNNT2 TNNT2 NEXN Full panel RBM20 TPM1 TGFB3 SCN5A MYBPC3 DSG2 MYH6 TNNI3 DSC2 TNNI3 MYL3 JUP TTN TTN RYR2 BAG3 MYLK2 TMEM43 DES Cardiomyopathy, familial DSP CRYAB EYA4 restrictive PKP2 Full panel Atrial fibrillation LAMA4 MYPN TNNI3 SGCD MYPN TNNT2 Full panel CSRP3 SCN5A MYBPC3 GJA5 TCAP ABCC9 ABCC9 SCN1B PLN SCN2B ACTC1 KCNQ1 MYH7 KCNE2 TMPO NPPA VCL NPPA TPM1 KCNA5 TNNC1 KCNJ2 GATAD1 4/1/2014 -

Minding the Calcium Store: Ryanodine Receptor Activation As a Convergent Mechanism of PCB Toxicity
Pharmacology & Therapeutics 125 (2010) 260–285 Contents lists available at ScienceDirect Pharmacology & Therapeutics journal homepage: www.elsevier.com/locate/pharmthera Associate Editor: Carey Pope Minding the calcium store: Ryanodine receptor activation as a convergent mechanism of PCB toxicity Isaac N. Pessah ⁎, Gennady Cherednichenko, Pamela J. Lein Department of Molecular Biosciences, School of Veterinary Medicine, University of California, Davis, CA 95616, USA article info abstract Keywords: Chronic low-level polychlorinated biphenyl (PCB) exposures remain a significant public health concern since Ryanodine receptor (RyR) results from epidemiological studies indicate that PCB burden is associated with immune system Calcium-induced calcium release dysfunction, cardiovascular disease, and impairment of the developing nervous system. Of these various Calcium regulation adverse health effects, developmental neurotoxicity has emerged as a particularly vulnerable endpoint in Polychlorinated biphenyls PCB toxicity. Arguably the most pervasive biological effects of PCBs could be mediated by their ability to alter Triclosan fi 2+ Bastadins the spatial and temporal delity of Ca signals through one or more receptor-mediated processes. This Polybrominated diphenylethers review will focus on our current knowledge of the structure and function of ryanodine receptors (RyRs) in Developmental neurotoxicity muscle and nerve cells and how PCBs and related non-coplanar structures alter these functions. The Activity dependent plasticity molecular and cellular mechanisms by which non-coplanar PCBs and related structures alter local and global Ca2+ signaling properties and the possible short and long-term consequences of these perturbations on neurodevelopment and neurodegeneration are reviewed. © 2009 Elsevier Inc. All rights reserved. Contents 1. Introduction ............................................... 260 2. Ryanodine receptor macromolecular complexes: significance to polychlorinated biphenyl-mediated Ca2+ dysregulation . -

Supp Table 1.Pdf
Upregulated genes in Hdac8 null cranial neural crest cells fold change Gene Symbol Gene Title 134.39 Stmn4 stathmin-like 4 46.05 Lhx1 LIM homeobox protein 1 31.45 Lect2 leukocyte cell-derived chemotaxin 2 31.09 Zfp108 zinc finger protein 108 27.74 0710007G10Rik RIKEN cDNA 0710007G10 gene 26.31 1700019O17Rik RIKEN cDNA 1700019O17 gene 25.72 Cyb561 Cytochrome b-561 25.35 Tsc22d1 TSC22 domain family, member 1 25.27 4921513I08Rik RIKEN cDNA 4921513I08 gene 24.58 Ofa oncofetal antigen 24.47 B230112I24Rik RIKEN cDNA B230112I24 gene 23.86 Uty ubiquitously transcribed tetratricopeptide repeat gene, Y chromosome 22.84 D8Ertd268e DNA segment, Chr 8, ERATO Doi 268, expressed 19.78 Dag1 Dystroglycan 1 19.74 Pkn1 protein kinase N1 18.64 Cts8 cathepsin 8 18.23 1500012D20Rik RIKEN cDNA 1500012D20 gene 18.09 Slc43a2 solute carrier family 43, member 2 17.17 Pcm1 Pericentriolar material 1 17.17 Prg2 proteoglycan 2, bone marrow 17.11 LOC671579 hypothetical protein LOC671579 17.11 Slco1a5 solute carrier organic anion transporter family, member 1a5 17.02 Fbxl7 F-box and leucine-rich repeat protein 7 17.02 Kcns2 K+ voltage-gated channel, subfamily S, 2 16.93 AW493845 Expressed sequence AW493845 16.12 1600014K23Rik RIKEN cDNA 1600014K23 gene 15.71 Cst8 cystatin 8 (cystatin-related epididymal spermatogenic) 15.68 4922502D21Rik RIKEN cDNA 4922502D21 gene 15.32 2810011L19Rik RIKEN cDNA 2810011L19 gene 15.08 Btbd9 BTB (POZ) domain containing 9 14.77 Hoxa11os homeo box A11, opposite strand transcript 14.74 Obp1a odorant binding protein Ia 14.72 ORF28 open reading -

A Comprehensive Analysis of the Expression of Crystallins in Mouse Retina Jinghua Xi Washington University School of Medicine in St
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2003 A comprehensive analysis of the expression of crystallins in mouse retina Jinghua Xi Washington University School of Medicine in St. Louis Rafal Farjo University of Michigan - Ann Arbor Shigeo Yoshida University of Michigan - Ann Arbor Timothy S. Kern Case Western Reserve University Anand Swaroop University of Michigan - Ann Arbor See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Xi, Jinghua; Farjo, Rafal; Yoshida, Shigeo; Kern, Timothy S.; Swaroop, Anand; and Andley, Usha P., ,"A comprehensive analysis of the expression of crystallins in mouse retina." Molecular Vision.9,. 410-419. (2003). https://digitalcommons.wustl.edu/open_access_pubs/1801 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Jinghua Xi, Rafal Farjo, Shigeo Yoshida, Timothy S. Kern, Anand Swaroop, and Usha P. Andley This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/1801 Molecular Vision 2003; 9:410-9 <http://www.molvis.org/molvis/v9/a53> © 2003 Molecular Vision Received 28 May 2003 | Accepted 19 August 2003 | Published 28 August 2003 A comprehensive analysis of the expression of crystallins in mouse retina Jinghua Xi,1 Rafal Farjo,3 Shigeo Yoshida,3 Timothy S. Kern,5 Anand Swaroop,3,4 Usha P. Andley1,2 Departments of 1Ophthalmology and Visual Sciences and 2Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. -

Next Generation Sequencing for Molecular Confirmation of Hereditary
Arch Cardiol Mex. 2015;85(1):68---72 www.elsevier.com.mx SPECIAL ARTICLE Next generation sequencing for molecular confirmation of hereditary sudden cardiac death syndromes a,∗ b b b Manlio F. Márquez , David Cruz-Robles , Selene Ines-Real , Gilberto Vargas-Alarcón , a Manuel Cárdenas a Departamento de Electrofisiología, Instituto Nacional de Cardiología Ignacio Chávez, México, D.F., Mexico b Departamento de Biología Molecular, Instituto Nacional de Cardiología Ignacio Chávez, México, D.F., Mexico Received 26 March 2014; accepted 8 December 2014 KEYWORDS Abstract Hereditary sudden cardiac death syndromes comprise a wide range of diseases result- Arrhythmias; ing from alteration in cardiac ion channels. Genes involved in these syndromes represent diverse Hereditary sudden mutations that cause the altered encoding of the diverse proteins constituting these channels, cardiac death thus affecting directly the currents of the corresponding ions. In the present article we will syndromes; briefly review how to arrive to a clinical diagnosis and we will present the results of molecular Right ventricle genetic studies made in Mexican subjects attending the SCD Syndromes Clinic of the National arrhythmogenic Institute of Cardiology of Mexico City. cardiomyopathy; © 2014 Instituto Nacional de Cardiología Ignacio Chávez. Published by Masson Doyma México Brugada syndrome S.A. All rights reserved. PALABRAS CLAVE Confirmación diagnóstica molecular mediante secuenciación masiva de nueva Arritmias; generación (‘‘next generation sequencing’’) en síndromes hereditarios de muerte Síndromes súbita cardíaca hereditarios de Resumen Los síndromes hereditarios de muerte súbita cardíaca comprenden una amplia gama muerte súbita; Displasia de enfermedades resultantes de la alteración en los canales iónicos cardíacos. Los genes implicados en estos síndromes presentan mutaciones que causan alteraciones de las diversas Arritmogénica del proteínas que constituyen estos canales y que, por lo tanto, afectan directamente a las difer- ventriculo derecho; entes corrientes iónicas. -

Related Macular Degeneration and Cutis Laxa
UvA-DARE (Digital Academic Repository) Genetic studies of age-related macular degeneration Baas, D.C. Publication date 2012 Document Version Final published version Link to publication Citation for published version (APA): Baas, D. C. (2012). Genetic studies of age-related macular degeneration. General rights It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other than for strictly personal, individual use, unless the work is under an open content license (like Creative Commons). Disclaimer/Complaints regulations If you believe that digital publication of certain material infringes any of your rights or (privacy) interests, please let the Library know, stating your reasons. In case of a legitimate complaint, the Library will make the material inaccessible and/or remove it from the website. Please Ask the Library: https://uba.uva.nl/en/contact, or a letter to: Library of the University of Amsterdam, Secretariat, Singel 425, 1012 WP Amsterdam, The Netherlands. You will be contacted as soon as possible. UvA-DARE is a service provided by the library of the University of Amsterdam (https://dare.uva.nl) Download date:05 Oct 2021 G������ S������ �� A��-������� M������ D����������� D����������� M������ G������ S������ �� A��-������� | 2012 D�������� C. B��� G������ S������ �� A��-������� M������ D����������� D�������� C. B��� cover.indd 1 31-10-12 08:36 Genetic Studies of Age-related Macular Degeneration Dominique C. Baas Chapter 0.indd 1 23-10-12 19:24 The research described in this thesis was conducted at the Netherlands Institute for Neuroscience (NIN), an institute of the Royal Netherlands Academy of Arts and Sciences, Department of Clinical and Molecular Ophthalmogenetics, Amsterdam, The Netherlands.