TARKA® (Trandolapril/Verapamil Hydrochloride ER Tablets)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Familial Mediterranean Fever
:: Familial Mediterranean fever – This document is a translation of the French recommendations drafted by Prof Gilles Grateau, Dr Véronique Hentgen, Dr Katia Stankovic Stojanovic and Dr Gilles Bagou reviewed and published by Orphanet in 2010. – Some of the procedures mentioned, particularly drug treatments, may not be validated in the country where you practice. Synonyms: Periodic disease, FMF Definition: Familial Mediterranean fever (FMF) is an auto-inflammatory disease of genetic origin, affecting Mediterranean populations and characterised by recurrent attacks of fever accompanied by polyserositis that causes the symptoms. Colchicine is the basic reference treatment and is designed to tackle inflammatory attacks and prevent amyloidosis, the most severe complication of FMF. Further information: See the Orphanet abstract http://www.orpha.net/data/patho/Pro/en/Emergency_FamilialMediterraneanFever-enPro920.pdf ©Orphanet UK 1/5 Pre-hospital emergency care recommendations Call for a patient suffering from Familial mediterranean fever Synonyms periodic disease FMF Mechanisms auto-inflammatory disease that affects Mediterranean populations in particular, due to mutation of the MEFV gene that codes pyrin or marenostrin, this being the underlying cause of congenital immune dysfunction; repeated inflammatory attacks can lead to amyloidosis, particularly renal Specific risks in emergency situations acute inflammatory attack, particularly abdominal (pseudo-surgical), but also thoracic, articular (knees) or testicular fever as an expression -

Guideline for Preoperative Medication Management
Guideline: Preoperative Medication Management Guideline for Preoperative Medication Management Purpose of Guideline: To provide guidance to physicians, advanced practice providers (APPs), pharmacists, and nurses regarding medication management in the preoperative setting. Background: Appropriate perioperative medication management is essential to ensure positive surgical outcomes and prevent medication misadventures.1 Results from a prospective analysis of 1,025 patients admitted to a general surgical unit concluded that patients on at least one medication for a chronic disease are 2.7 times more likely to experience surgical complications compared with those not taking any medications. As the aging population requires more medication use and the availability of various nonprescription medications continues to increase, so does the risk of polypharmacy and the need for perioperative medication guidance.2 There are no well-designed trials to support evidence-based recommendations for perioperative medication management; however, general principles and best practice approaches are available. General considerations for perioperative medication management include a thorough medication history, understanding of the medication pharmacokinetics and potential for withdrawal symptoms, understanding the risks associated with the surgical procedure and the risks of medication discontinuation based on the intended indication. Clinical judgement must be exercised, especially if medication pharmacokinetics are not predictable or there are significant risks associated with inappropriate medication withdrawal (eg, tolerance) or continuation (eg, postsurgical infection).2 Clinical Assessment: Prior to instructing the patient on preoperative medication management, completion of a thorough medication history is recommended – including all information on prescription medications, over-the-counter medications, “as needed” medications, vitamins, supplements, and herbal medications. Allergies should also be verified and documented. -

The Effect of Caffeine and Trifluralin on Chromosome Doubling in Wheat
plants Article The Effect of Caffeine and Trifluralin on Chromosome Doubling in Wheat Anther Culture Sue Broughton *, Marieclaire Castello, Li Liu, Julie Killen, Anna Hepworth and Rebecca O’Leary Department of Primary Industries and Regional Development, 3 Baron-Hay Court, South Perth 6151, Western Australia, Australia; [email protected] (M.C.); [email protected] (L.L.); [email protected] (J.K.); [email protected] (A.H.); Rebecca.O'[email protected] (R.O.) * Correspondence: [email protected] Received: 3 December 2019; Accepted: 10 January 2020; Published: 15 January 2020 Abstract: Challenges for wheat doubled haploid (DH) production using anther culture include genotype variability in green plant regeneration and spontaneous chromosome doubling. The frequency of chromosome doubling in our program can vary from 14% to 80%. Caffeine or trifluralin was applied at the start of the induction phase to improve early genome doubling. Caffeine treatment at 0.5 mM for 24 h significantly improved green plant production in two of the six spring wheat crosses but had no effect on the other crosses. The improvements were observed in Trojan/Havoc and Lancer/LPB14-0392, where green plant numbers increased by 14% and 27% to 161 and 42 green plants per 30 anthers, respectively. Caffeine had no significant effect on chromosome doubling, despite a higher frequency of doubling in several caffeine treatments in the first experiment (67–68%) compared to the control (56%). In contrast, trifluralin significantly improved doubling following a 48 h treatment, from 38% in the control to 51% and 53% in the 1 µM and 3 µM trifluralin treatments, respectively. -

Your 2017 Three-Tier
Your 2017 Three-Tier Prescription Drug List effective January 1, 2017 Connecticut Advantage Three-Tier Please read: This document contains information about commonly prescribed medications. This Prescription Drug List (PDL) is accurate as of January 1, 2017 and is subject to change after this date. The next anticipated update will be in July 2017. Your estimated coverage and copay/co-insurance may vary based on the benefit plan you choose and the effective date of the plan. For additional information: Call the toll-free member phone number on your health plan ID card. Visit myuhc.com® • Locate a participating retail pharmacy by ZIP code. • Look up possible lower-cost medication alternatives. • Compare medication pricing and options. 1 Your Prescription Drug List This Prescription Drug List (PDL) outlines covered medications for certain conditions and organizes them into cost levels, also known as tiers. An important part of the PDL is giving you choices so you and your doctor can choose the best course of treatment for you. Go to myuhc.com® for complete drug information Since the PDL may change, we encourage you to visit our website, myuhc.com. This website is the best source for up-to-date information about the medications your pharmacy benefit covers, possible lower-cost options, and cost comparisons. To view PDL information without logging on to the member portal, visit myuhc.com and select “Pharmacy Information” under “Links and Tools.” 2 At UnitedHealthcare, we want to help you better understand your medication options. Your pharmacy benefit offers flexibility and choice in determining the right medication for you. -
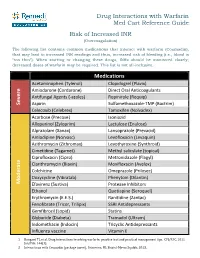
Drug Interactions with Warfarin Med Cart Reference Guide Risk of Increased INR (Overcoagulation)
Drug Interactions with Warfarin Med Cart Reference Guide Risk of Increased INR (Overcoagulation) The following list contains common medications that interact with warfarin (Coumadin), that may lead to increased INR readings and thus, increased risk of bleeding (i.e., blood is “too thin”). When starting or changing these drugs, INRs should be monitored closely; decreased doses of warfarin may be required. This list is not all-inclusive. Medications Acetaminophen (Tylenol) Clopidogrel (Plavix) Amiodarone (Cordarone) Direct Oral Anticoagulants Antifungal Agents (‐azoles) Ropinirole (Requip) Severe Aspirin Sulfamethoxazole‐TMP (Bactrim) Celecoxib (Celebrex) Tamoxifen (Nolvadex) Acarbose (Precose) Isoniazid Allopurinol (Zyloprim) Lactulose (Enulose) Alprazolam (Xanax) Lansoprazole (Prevacid) Amlodipine (Norvasc) Levofloxacin (Levaquin) Azithromycin (Zithromax) Levothyroxine (Synthroid) Cimetidine (Tagamet) Methyl salicylate (topical) Ciprofloxacin (Cipro) Metronidazole (Flagyl) Clarithromycin (Biaxin) Moxifloxacin (Avelox) Colchicine Omeprazole (Prilosec) Doxycycline (Vibratab) Phenytoin (Dilantin) Moderate Efavirenz (Sustiva) Protease Inhibitors Ethanol Quetiapine (Seroquel) Erythromycin (E.E.S.) Ranitidine (Zantac) Fenofibrate (Tricor, Trilipix) SSRI Antidepressants Gemfibrozil (Lopid) Statins Glyburide (Diabeta) Tramadol (Ultram) Indomethacin (Indocin) Tricyclic Antidepressants Influenza vaccine Vitamin E 1. Bungard TJ, et al. Drug interactions involving warfarin: practice tool and practical management tips. CPJ/RPC. 2011 Jan/Feb. 144(1). 2. Interactions with Coumadin [package insert]. Princeton, NJ: Bristol‐Myers Squibb; 2013. Drug Interactions with Warfarin Med Cart Reference Guide Risk of Decreased INR (Undercoagulation) The following list contains common medications that interact with warfarin (Coumadin), which may lead to decreased INR readings and thus, increased risk of clotting, strokes, and DVTs (i.e., blood is “not thin enough”). When starting or changing these drugs, INRs should be monitored closely; increased doses of warfarin may be required. -

Drugs Which Can Affect Near Vision: a Useful List
Drugs Which Can Affect Near Vision: A Useful List Joanne L. Smith B.Sc., Ph.Phm.* J. Raymond Buncic, M.D., F.R.C.S.(C)t ABSTRACT This paper documents a list of drugs that cause problems with near vision, by virtue of effects on accommodation, occasionally refractive error and diplopia. It is meant as a reference aid to the clinician when confronted with problems of focusing on near objects or print. There are many drugs that have been reported to interfere with near or reading vision, producing blurring, decreased accommodation and diplopia. This paper lists the drugs that have been reported in the literature to produce symptoms which interfere with near vision. Case reports for the listed drugs vary greatly from many to few. The drugs have been divided into the following categories: those causing (A) blurring at near, (B) diplopia and (C) induced myopia. Those drugs which only rarely cause these symptoms have been omitted. From the Departments of Pharmacy* and Ophthalmologyt, The Hospital For Sick Children, Toronto, Ontario, Canada Requests for reprints should be addressed to: Dr. J. Raymond Buncic, Department of Ophthalmology, The Hospital For Sick Children, 555 University Ave., Toronto, Ontario, Canada M5G lX8 TABLE 1 DRUGS COMMONLY CAUSING DIFFICULTY WITH FOCUSING AT NEAR OR BLURRED VISION. DRUG INCIDENCE REFERENCE Antipsychotics Chlorpromazine 14-23 8 Clozapine 5 8,14 Fluphenazine 1.2-4.3 8 Haloperidol 6.8-16 8 Loxapine 12,14 Perphenazine 7.4-17.8 8 Pimozide 20 8 Risperidone 1-2%, >/= 10% 11 Thioridazine 0.6-18 8 Thiothixene 20 8 -

Life-Threatening Colchicine Drug Interactions John R
drugINTERACTIONS: insights and observations Life-threatening Colchicine Drug Interactions John R. Horn, PharmD, FCCP,and Philip D. Hansten, PharmD after starting clarithromycin. He subse- Table Drs. Horn and Hansten are both pro- quently developed multiorgan failure Selected Drugs That Inhibit fessors of pharmacy at the University and died.1 Another patient on col- of Washington School of Pharmacy. P-glycoprotein chicine developed fever, diarrhea, and For an electronic version of this arti- Amiodarone Nicardipine abdominal pain 3 days after starting cle, including references if any, visit Clarithromycin Propafenone clarithromycin.2 He went on to develop www.hanstenandhorn.com. Cyclosporine Quinidine dehydration, pancytopenia, and acido- Diltiazem Ritonavir sis, but he survived. Two other patients Erythromycin Saquinavir ew drugs have been around as on colchicine died from agranulocyto- Indinavir Tacrolimus long as colchicine—it was men- sis when clarithromycin was given con- Itraconazole Tamoxifen tioned at least as far back as currently.3 F Ketoconazole Verapamil the time of Nero. Colchicine can be Similar life-threatening reactions Nelfinavir very effective in the treatment of gout have been reported in patients on and familial Mediterranean fever. Un- colchicine who received concomitant fortunately, however, its therapeutic therapy with other PGP inhibitors.4-7 In Managing Interactions effects (primarily on white blood cells) most cases, colchicine toxicity has Colchicine should not be used with can lead to life-threatening toxicity if occurred in patients on chronic clarithromycin or erythromycin, and colchicine plasma concentrations be- colchicine therapy who are then start- given the potential for fatal outcomes, come too high. One of the causes of ed on a PGP inhibitor. -

Biology, Breeding and Applications of Cannabis Sativa
The Cream of the Crop: Biology, Breeding and Applications of Cannabis sativa Susanne Schilling*1; CarolineA:Dowling ∗ 1; JiaqiShi ∗ 1; LouiseRyan1; DavidHunt1; EveOReilly1; AntoinetteS:P erry1; OliverKinnane1; P aulF:McCabe1; andRainerMelzer1 1University College Dublin October 1, 2020 Abstract Cannabis sativa is an extraordinarily versatile species. Hemp and its cousin marijuana, both C. sativa, have been used for millennia as a source of fibre, oil and for medicinal, spiritual and recreational purposes. Because the consumption of Cannabis can have psychoactive effects, the plant has been widely banned throughout the last century. In the past decade, evidence of its medicinal properties did lead to the relaxation of legislation in many countries around the world. Consequently, the genetics and development of Cannabis as well as Cannabis-derived products are the subject of renewed attention.Here, we review the biology of C. sativa, including recent insights from taxonomy, morphology and genomics, with an emphasis on the genetics of cannabinoid synthesis. Because the female Cannabis flower is of special interest as the site of cannabinoid synthesis, we explore flower development, flowering time well as the species’ unique sex determination system in detail. Furthermore, we outline the tremendous medicinal, engineering, and environmental opportunities that Cannabis bears. Together, the picture emerges that our understanding of Cannabis biology currently progresses at an unusual speed. A future challenge will be to preserve the multi-purpose nature of Cannabis, and to harness its medicinal properties and sustainability advantages simultaneously. *: These authors contributed equally to this work and should be considered joint first authors ˆ: for correspondence: susanne.schilling [at] udc.ie; rainer.melzer[at]ucd.ie Keywords: building materials, Cannabis sativa, cannabinoids, crop breeding, flower development, flowering time, hemp, marijuana, sex determination, sustainability 1. -

Tetraparesis Associated with Colchicine Is Probably Due To
Papers BMJ: first published as 10.1136/bmj.38568.639688.F7 on 7 September 2005. Downloaded from Tetraparesis associated with colchicine is probably due to inhibition by verapamil of the P-glycoprotein efflux pump in the blood-brain barrier Uwe Tröger, Hartmut Lins, Jean-M Scherrmann, Claus-Werner Wallesch, Stefanie M Bode-Böger An 83 year old man had an acute attack of gout. He Institute of Clinical treated himself with colchicine drops (2 mg in two days) Colchicine concentration in cerebrospinal fluid Pharmacology, Otto-von-Guericke- and received diclofenac because of continuous pain. Con- Verapamil concentration in serum Colchicine concentration in serum University, currently he had muscle weakness in his limbs. Four days Leipziger Strasse Norverapamil concentration in serum 44, D-39120 later he became immobile and was transferred to hospital 2.0 with flaccid tetraparesis (British Medical Research Council Magdeburg, Germany grade II-III). He had no signs of infection, hepatic or renal 1.6 Uwe Tröger impairments, or stroke. Laboratory values were normal 500 400 consultant in clinical except a brief increase of creatine kinase to 1288.2 IU/l. 1.2 300 pharmacology Repeated nerve conduction studies did not show any 200 Stefanie M 0.8 100 Bode-Böger relevant pathology, including normal terminal latencies concentration (ng/ml) and F wave latencies. Electromyography subsequently 0 Verapamil/norverapamil professor in clinical pharmacology showed signs of lower motor neurone lesion. Muscle 0.4 Colchicine intake 1 mg/day biopsy showed one isolated vacuole that could not be Colchicine concentration (ng/ml) Department of 0 Neurology, related to a colchicine induced myoneuropathy. -

American Rheumatism Association
Ann Rheum Dis: first published as 10.1136/ard.14.4.412 on 1 December 1955. Downloaded from Ann. rheum. Dis. (1955), 14, 412. AMERICAN RHEUMATISM ASSOCIATION PROCEEDINGS OF THE ANNUAL MEETING, 1955 The annual meeting of the American Rheumatism brane were prepared by freezing and thawing in distilled Association was held at Atlantic City, New Jersey, water. The substrate was a viscous mucoprotein con- on June 3 and 4, 1955, under the presidency of taining 75 per cent. chondroitin sulphate and 25 per cent. Dr. Edward W. Boland of Los Angeles. Abstracts of protein, isolated from cartilage. A rapid fall in the viscosity of the mucoprotein was observed, with extracts thirty papers and the discussions thereon are printed both of leucocytes and ofrheumatoid synovial membrane. below. One session was arranged in conjunction The fall in viscosity paralleled that obtained with crystal- with the American Council on Rheumatic Fever and line trypsin. The amount of fall in viscosity was propor- Congenital Heart Disease. Dr. Boland's presi- tional to the concentration of added extract, and was not dential address formed part of the panel discussion changed by adding reducing agents or a variety of on "New Analogues of Adrenocortical Steroids" proteins. The activity of the extracts showed a maxi- (Moderator: Dr. J. J. Bunim). A second panel mumppH between 5*2 and 6 5; the temperature coefficient discussion was held on "Reconstructive Surgery in of the reaction was 2- 5. Heating the extracts rendered Arthritis" (Moderator: Dr. Albert J. Key). them inactive. Soya bean inhibitor blocked the effect of trypsin, but not that of the leucocyte or synovial mem- brane extracts. -

The Validation of a 15 STR Multiplex PCR for Cannabis Species
Int J Legal Med (2012) 126:601–606 DOI 10.1007/s00414-012-0706-6 ORIGINAL ARTICLE The validation of a 15 STR multiplex PCR for Cannabis species Stephan Köhnemann & Johanna Nedele & Daniela Schwotzer & Julia Morzfeld & Heidi Pfeiffer Received: 31 January 2012 /Accepted: 26 April 2012 /Published online: 10 May 2012 # Springer-Verlag 2012 Abstract Trade and acquisition of Cannabis drugs are illegal and are therefore prohibited by law in many countries [2]. in many countries worldwide; nevertheless, crimes related The main psychoactive compound of drug Cannabis is Δ9- with these drugs are a major problem for the investigative tetrahydrocannabinol (THC) which can be found in high authorities. With this manuscript, we want to introduce a 15 concentrations in leaves and inflorescences. short tandem repeat (STR) Cannabis marker set that can be Techniques to distinguish Cannabis on the DNA level amplified in one PCR reaction. This multiplex PCR is specific were developed previously by different working groups to Cannabis species and combines highly informative STR [3–7]. Short tandem repeats (STRs) appear to have the best markers.The15STRmultiplexiseasytouseandwas discrimination ability; nevertheless, none of these groups validated according to common laboratory quality standards. tried to combine more than six STRs in one multiplex Due to the fact that a lot of Cannabis plants are cultivated by PCR. STRs are repetitive sequences of up to six bases at a clonal propagation and may show aneuploidy, polyploidy or defined gene locus, found normally in the non-coding re- multiple gene loci, it is not possible to apply biostatistics that gion of autosomal or gonosomal DNA. -

483 Administrative Records, Texas Department of Criminal Justice
Consistent with the terms of the Court’s May 22, 2017 scheduling order, the record has been redacted for all information that plaintiff, Texas Department of Criminal Justice (Texas), has identified as confidential. In addition, Defendants have also redacted information that the drug’s supplier and broker have separately advised the agency they consider confidential and private, as well as information the agency itself generally treats as confidential. This information has been redacted pending final FDA’s review of confidentiality claims, and our filing of the record with these redactions does not necessarily reflect our agreement with all of the claims of confidentiality Defendants have received. Defendants explicitly reserve the right to make an independent determination regarding the proper scope of redactions at a later time. Should we identify any of Texas’s redactions that are over-broad or otherwise improper, we will work with Texas’s counsel to revise the redactions in the record. Exhibit 14 FDA 095 Case 1:11-cv-00289-RJL Document 13-3 Filed 04/20/11 Page 1 of 74 CERTJFICATE Pursuant to the provisions ofRule 44 ofthe Federal Rules ofCivil Procedure, I hereby certifY that John Verbeten, Director ofthe Operations and Policy Branch, Division of Import Operations and Policy, Office ofRegional Operations, Office ofRegulatory Affairs, United States Food and Drug Administration, whose declaration is attached, has custody ofofficial records ofthe United States Food and Drug Administration. In witness whereof, I have, pursuant to the provision ofTitle 42, United States Code, Section 3505, and FDA StaffManual Guide 1410.23, hereto set my hand and caused the seal ofthe Department ofHealth and Human Services to be affixed th.is ~oi{.