Studies on Tin Compounds and Complfies of the Platinum
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Characterization of Strychnine Binding Sites in the Rodent
CHARACTERIZATION OF STRYCHNINE BINDING SITES IN THE RODENT SPINAL CORD BY VINCENT MAURICE O’CONNOR UNIVERSITY COLLEGE LONDON A thesis submitted for the degree of Doctor of Philosophy from the University of London, 1992. I ProQuest Number: 10608898 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 10608898 Published by ProQuest LLC(2017). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 This thesis is dedicated to the select band o f people, including the most recent arrivals and the sorely missed departed, that I hold closest in my affections. "At the end of the day" they make It all worthwhile. Remember, in the words of the famous actor whose name at present escapes me; "Nobody said it would be easy". Although my own feeling is that Nobody was probably wrong. II Thesis abstract The convulsant alkaloid strychnine is a selective and highly potent antagonist at postsynaptic receptor for the inhibitory neurotransmitter glycine . These properties have led to the extensive use of strychnine as a ligand to probe the postsynaptic glycine receptor. Despite the recent increased understanding of the molecular structure of this receptor protein there is still much dispute as to the nature of the interaction between glycine and strychnine. -

Chemical Resistance of Plastics
(c) Bürkle GmbH 2010 Important Important information The tables “Chemical resistance of plastics”, “Plastics and their properties” and “Viscosity of liquids" as well as the information about chemical resistance given in the particular product descriptions have been drawn up based on information provided by various raw material manufacturers. These values are based solely on laboratory tests with raw materials. Plastic components produced from these raw materials are frequently subject to influences that cannot be recognized in laboratory tests (temperature, pressure, material stress, effects of chemicals, construction features, etc.). For this reason the values given are only to be regarded as being guidelines. In critical cases it is essential that a test is carried out first. No legal claims can be derived from this information; nor do we accept any liability for it. A knowledge of the chemical and mechanical Copyright This table has been published and updated by Bürkle GmbH, D-79415 Bad Bellingen as a work of reference. This Copyright clause must not be removed. The table may be freely passed on and copied, provided that Extensions, additions and translations If your own experiences with materials and media could be used to extend this table then we would be pleased to receive any additional information. Please send an E-Mail to [email protected]. We would also like to receive translations into other languages. Please visit our website at http://www.buerkle.de from time to Thanks Our special thanks to Franz Kass ([email protected]), who has completed and extended these lists with great enthusiasm and his excellent specialist knowledge. -

Loba Chemie Pvt
TABLE OF CONTENT INTRODUCTION Contact US ii MD Letter iii Company Profile iv Latest Information v Label vi Certifications vii GMP Compliant Pharma Facility viii QC Capability ix R&D x Logistic xi COA xii SDS xiii Packing xiv Quality xvi Terms Of Sales xvii Application xviii Product Highlights xix Nanopowder & Carbon Nanotubes xxvii List of New Products xxviii ALPHABETICAL PRODUCT LISTING Price List Chemical 01-155 Ecosafe Safety Products 156-160 Macherey-Nagel Filtration Products 161-181 [email protected] I CONTACT US Contact us for more information on any of our products and services HEAD OFFICE - MUMBAI Loba Chemie Pvt. Ltd., Jehangir Villa, 107, Wode House Road, Colaba, Mumbai-400005 Maharashtra, India Tel: +91(022) 6663 6663 Fax: +91(022) 6663 6699 MANUFACTURING UNIT (TARAPUR) Loba Chemie Pvt. Ltd., Plot No.: D-22, M.I.D.C. Tarapur, Boisar, Taluka: Palghar, Thane-401506 Maharashtra, India Ph: +91(02525) 300 001 Stay up to date about our range and availability www.lobachemie.com Get in touch with us General Enquiry: [email protected] Technical Query: [email protected] Domestic Sales: [email protected] Export Sales: [email protected] Follow us on: ii www.lobachemie.com WELCOME AT LOBA CHEMIE Dear Valued Reader Another exciting year has gone by and we would like to start the New Year, as usual, with a new catalogue featuring more innova- tive highest quality range of routine and novel Laboratory Chemi- cal and Fine Reagents. With our more than 4 decads of expertise in Laboratory Chemical and Fine Reagents we not only bring you a complete Laboratory at your fingertips but in addition with our expertise we have palced our brand of products in a very competitive position for years to come. -

|||||III USOO5547541A United States Patent (19) 11) Patent Number: 5,547,541 Hansen Et Al
|||||III USOO5547541A United States Patent (19) 11) Patent Number: 5,547,541 Hansen et al. 45) Date of Patent: Aug. 20, 1996 54) METHOD FOR DENSEFYING FIBERS USING OTHER PUBLICATIONS A DENSIFYINGAGENT Gugliemelli et al., "Base-Hydrolyzed Starch-Polyacryloni (75) Inventors: Michael R. Hansen, Seattle; Richard trile (S-PAN) Graft Copolymer. S-PAN-1:1, PAN. M. W. H. Young, Sr., Renton, both of Wash. 794,000*", J. of Applied Copolymer Science, 13:2007-2017 (1969). (73) Assignee: Weyerhaeuser Company, Federal Way, Weaver et al., "Hydrolyzed Starch-Polyacrylonitrile Graft Wash. Copolymers: Effect of Structure on Properties', J. of Applied Polymer Science, 15:3015–3024 (1971). Weaver et al., "Highly Absorbent Starch-Based Polymer." 21 Appl. No.: 197483 Northern Regional Research Laboratory, Agricultural 22) Filed: Feb. 16, 1994 Research Service, U.S. Dept. of Agriculture, Peoria, Illinois, pp. 169-177. Related U.S. Application Data "Super slurpers: Time for change?,” Chemical Week, pp. 21-22 (Jul. 24, 1974). (63) Continuation-in-part of Ser. No. 931,059, Aug. 17, 1992, S. Lammie, "Use of Glycerine as a Softener for Paper Ser. No. 931,277, Aug. 17, 1992, Ser. No. 931,213, Aug. 17, 1992, Pat. No. 5,300,192, Ser. No. 931,278, Aug. 17, 1992, Products." The World's Paper Trade Review, Dec. 13, 1962, Pat. No. 5,352,480, Ser. No. 931,284, Aug. 17, 1992, Pat. p. 2050. No. 5,308,896, Ser. No. 931,279, Aug. 17, 1992, Ser. No. Lindsay, "Absorbent Starch Based Co-polymers- Their 107,469, Aug. 17, 1993, Ser. No. 108,219, Aug. -

Spectra Library Index Ichem/SDBS Raman Library
Ichem / SDBS Raman スタンダードライブラリー 株式会社 エス・ティ・ジャパン S p e c t r a L i b r a r y I n d e x (Ver. 40) Ichem / SDBS Raman Library ライブラリー名:ラマンスタンダードライブラリー 商品番号:60001-40 株式会社 エス・ティ・ジャパン 〒103-0014 東京都中央区日本橋蛎殻町 1-14-10 Tel: 03-3666-2561 Fax:03-3666-2658 http://www.stjapan.co.jp 1 【販売代理店】(株)テクノサイエンス Tel:043-206-0155 Fax:043-206-0188 https://www.techno-lab-co.jp/ Ichem / SDBS Raman スタンダードライブラリー 株式会社 エス・ティ・ジャパン ((1,2-DIETHYLETHYLENE)BIS(P-PHENYLENE))DIACETATE ((2-(3-BENZYLSULFONYL-4-METHYLCYCLOHEXYL)PROPYL)SULFONYLMETHYL)BENZENE ((2,4,6-TRIOXOHEXAHYDRO-5-PYRIMIDINYL)IMINO)DIACETIC ACID ((2-CARBOXYETHYL)IMINO)DIACETIC ACID ((2-HYDROXYETHYL)IMINO)DIACETIC ACID ((2-NITROBENZYL)IMINO)DIACETIC ACID ((2-SULFOETHYL)IMINO)DIACETIC ACID ((3-(1-BROMO-1-METHYLETHYL)-7-OXO-1,3,5-CYCLOHEPTATRIEN-1-YL)OXY)DIFLUOROBORANE ((N-BENZYLOXYCARBONYL-L-ISOLEUCYL)-L-PROLYL-L-PHENYLALANYL)-N(OMEGA)-NITRO-L-ARGININE 4-NITROBENZYL ESTER (-)-2-AMINO-1-BUTANOL (-)-2-AMINO-6-MERCAPTOPURINE RIBOSIDE (-)-6,8-P-MENTHADIEN-2-OL (-)-DIACETYL-L-TARTARIC ACID (-)-DIBENZOYL-L-TARTARIC ACID (-)-DI-P-ANISOYL-L-TARTARIC ACID (-)-DI-P-TOLUOYL-L-TARTARIC ACID (-)-KAURENE (-)-MENTHOL (-)-MYRTENOL (-)-N,N,N',N'-TETRAMETHYL-D-TARTARDIAMIDE (-)-N,N'-DIBENZYL-D-TARTRAMIDE (+)-1,3,3-TRIMETHYLNORBORNANE-2-ONE (+)-2-(2,4,5,7-TETRANITRO-9-FLUORENYLIDENEAMINOOXY)PROPIONIC ACID (+)-2-AMINO-1-BUTANOL (+)-2-PINENE (+)-3,9-DIBROMOCAMPHOR (+)-3-CARENE (+)-5-BROMO-2'-DEOXYURIDINE (+)-AMMONIUM 3-BROMO-8-CAMPHORSULFONATE (+)-CAMPHOR (+)-CAMPHOR OXIME (+)-CAMPHORIC ACID (+)-CATECHIN (+)-DI-P-ANISOYL-D-TARTARIC -

Aspects of the Radiation Chemistry and Protection of Peptides
A S P E C T S 0 F T HE R A D I A T I O N C H E M I S T R Y A, N D P R O T E C T I O N O F P E P T I D E S A thesis submitted in partial fulfillment of the requirements for the degree of M A S T E R 0 F S C I E N C E in the University of New South Wales by J.A. HOURIGAN, B.Sc. July, 1971. (11) The work contained in this thesis has not been submitted for a degree or similar award to any other University or Institution. J.A. HOURIGAN (111) Aspects of the radiation chellliatry and protection of the polypeptide chain of catalase were investigated, employing the soluble enz,111le and also insoluble, poly(diazostyrene)-bound catalase. Prior investigation of the structure of the inaolubilized catalase by ion-exchange chromatography of its hydrolysate showed that approximately 140 residues (histidine, lysine, cysteine and tyrosine, i.e. roughly half the total number of these residues per catalase aolecule) were covalently linked to poly(diazostyrene) molecules. The conjugate was pictured as a catalase molecule to which a large number (20-100) of poly(diazostyrene) molecules were covalently linked. The results indicate further, and in agreement with the results of irradiation experiments, that the tyrosine and arginine residues are mainly located in the interior of the active conformer of the catalase molecule. A novel pyrolysis-gas chromatographic technique was developed for the estimation of the protein content of poly(diazostyrene)-bound catalase. -
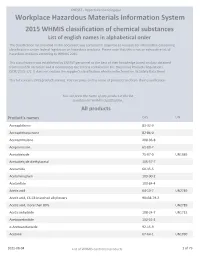
2015 WHMIS Classification : List of English Names in Alphabetical Order
CNESST - Répertoire toxicologique Workplace Hazardous Materials Information System 2015 WHMIS classification of chemical substances List of english names in alphabetical order The classification list provided in this document was compiled in response to requests for information concerning classifications under federal legislation on hazardous products. Please note that this is not an exhaustive list of hazardous products according to WHMIS 2015. This classification was established by CNESST personnel to the best of their knowledge based on data obtained from scientific literature and it incorporates the criteria contained in the Hazardous Products Regulations (SOR/2015-17). It does not replace the supplier's classification which can be found on its Safety Data Sheet. This list contains 2425 products names. You can press on the name of products to obtain their classification. You can press the name of any product in the list to obtain his WHMIS classification. All products Product's names CAS UN Acenaphthene 83-32-9 Acenaphthoquinone 82-86-0 Acenaphthylene 208-96-8 Acepromazine 61-00-7 Acetaldehyde 75-07-0 UN1089 Acetaldehyde diethylacetal 105-57-7 Acetamide 60-35-5 Acetaminophen 103-90-2 Acetanilide 103-84-4 Acetic acid 64-19-7 UN2789 Acetic acid, C6-C8 branched alkyl esters 90438-79-2 Acetic acid, more than 80% UN2789 Acetic anhydride 108-24-7 UN1715 Acetoacetanilide 102-01-2 o-Acetoacetaniside 92-15-9 Acetone 67-64-1 UN1090 2021-08-04 List of WHMIS controlled products 1 of 79 CNESST - Répertoire toxicologique Acetonitrile 75-05-8 UN1648 -

Hhe Synthesis of Compounds Related to Colchicine a Thesis Submitted For
Hhe Synthesis of Compounds Related to Colchicine A thesis submitted for the degree of Doctor of Philosophy at the University of Glasgow George. 1. Buchanan, B.Sc, Department of Chemistry University of Glasgow. July 1954. ProQuest Number: 13838739 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 13838739 Published by ProQuest LLC(2019). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 Preface. The author wishes to express his gratitude to Professor J. W. Cook, P.P.S., and Dr. J. D. Loudon, for their guidance, and advice in the work described. He is also indebted to the Carnegie Trust for the Universities of Scotland, for the award of a Research Scholarship. Because of war-time demands, this scholarship was held for only one year, and covers Part 1 of this thesis. There is an interval of about four years between the work described in Parts I and II, and this has necessitated a somewhat lengthy Historical Introduction to Part II. The micro-analyses, unless otherwise acknowledged were carried out by Mr. JVM . L. -

Molecular Characterization of a Non-Glycinergic Strychnine Binding
MOLECULAR CHARACTERIZATION OF A NON GLYCEVERGIC STRYCHNINE BINDING SITE IN THE RODENT SPINAL CORD BY HANQEVG GUO HOLMAN UNIVERSITY COLLEGE LONDON Thesis submitted for the degree of Doctor of Philosophy from the University of London, 1996. ProQuest Number: 10045733 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest. ProQuest 10045733 Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 Dedication % ^ # # # # * To my parents: for all the years of support and encouragement they have given me. II Acknowledgem ents I am pleased to have this opportunity to thank Dr. Jonathan Fry for his excellent supervision and advice throughout this project. This work would not have been possible without the co-operation of others. Special thanks to Dr.S.Moss for general help and advice along the project; Professor R. Mir sky, Department of Anatomy and Development Biology, UCL for supplying the antibodies; Mr. N. Totty and Dr. J. Hsuan, Ludwig Institute for Cancer Research, London for sequencing the peptide. I wish to acknowledge the help of Ms. B. Collins and Dr. C. Williams. -

Dictionaire Des Termes De Médicine
Digitized by the Internet Archive in 2008 with funding from IVIicrosoft Corporation http://www.archive.org/details/dictionaryofmedi02mruoft FRANgAIS-ANGLAIS. : ^^(ii^^ DICTIONSAIEE TEEMES DE MEDECINE. FT? A.lSrCAIS - AISTG^LAIS. PAR H. DE MEEIC, Mgmbbe du College Royal des Chiruhoiens d'Anoletbrre ; Chirurgien DE L'HdPiTAL Pran^ais X LONDRES; Membke DE la'Soci£ti5 d'Hygikne DE Paris ; Membre Correspondant de LA SOCIETE DE DeRMATOLOOIE ET DE SyPHILIORAPHIE DK FARIS, ETC. LONDON BAILLIERE, TINDALL AND COX, 20 & 21, KING WILLIAM STREET, STRAND. 1899. N/2, ; PEEFACE. En redigeant un dictionnaire fran^ais-anglais des termes de medecine, il est bien difficile de se borner aux mots qui se rapportent uniquement a la medecine, a la chirurgie, a I'anatomie, a la pathologie, etc. Meme Ton m'a reproche, lors de la publication de mon dictionnaire anglais-fran(;ai8, d'avoir insere des mots de langage courant qui, a premiere vue, paraissaient n'avoir rien a faire dans un tel ouvrage pourtant j'ai cru bien faire, vu le grand nombre de fois qu'on les rencontre dans les livres de medecine, de conserver dans cette seconde partie beaucoup de termes que Ton pourrait peut-etre inclure dans cette categorie. La seconde partie est moins longue que la premiere puisqu'il est inutile de mettre les genres aux mots anglais et parce que je n'ai pas juge necessaire de repeter les traitements en medecine ni les operations de chirurgie que Ton trouvera dans la premiere partie sous les noms des medecins et des chirurgiens qui les ont d'abord pratique's. -

Aldrich FT-IR Collection Edition II
Aldrich FT-IR Collection Edition II Library Listing – 18,454 spectra This library represents the most comprehensive collection of FT-IR spectral references available. It contains the most common chemicals found in the Aldrich Handbook of Fine Chemicals. The Aldrich Collection of FT-IR Spectra Edition II library contains spectra of 18,454 pure compounds and comes with the Second Edition of the Aldrich Library of FT-IR Spectra (a 3-volume book set). All spectra were acquired by Sigma-Aldrich Co. and were processed by Thermo Electron Corporation. Solid samples were run as mulls. Liquids were run neat. The following information is available when known: • Index number • Compound name • Molecular formula • Molecular weight • Boiling point • Melting point • Flash point • Density • Refractive index • Category name This library represents the largest collection of spectra collected entirely by FT-IR instrumentation. Eight smaller Aldrich Material Specific Sub-Libraries are also available. Aldrich FT-IR Collection Edition II Index Compound Name Index Compound Name 14261 4,4'-Diphenyl-2,2'-dipyridyl 16353 (+)-5alpha-Androstan-17beta-ol-3-one 5936 ((1R)-(endo,anti))-(+)-3- benzoate, 98% Bromocamphor-8- sulfonic acid, 5260 (+)-6-Aminopenicillanic acid, 96% ammonium salt 10157 (+)-6-Methoxy-alpha-methyl-2- 2683 ((1R)-endo)-(+)-3-Bromocamphor, 98% naphthaleneacetic acid, 98% 5937 ((1S)-(endo,anti))-(-)-3-Bromocamphor- 16306 (+)-Androsta-1,4-diene-3,17-dione, 98% 8- sulfonic acid, ammonium salt 1148 (+)-Arabinogalactan 2684 ((1S)-endo)-(-)-3-Bromocamphor, -

September 1, 2000
1740 Technology Drive 408 441-0876 Suite 300 408 436-9306 San Jose, CA 95110 www.keenan.com License No. 0451271 June 10, 2014 Rachel Chow-Lucas Facilities Planner SAN JOSE/EVERGREEN COMMUNITY COLLEGE DISTRICT (CCD) 4750 San Felipe Road San Jose, CA 95135 RE: SAN JOSE CITY COLLEGE AND EVERGREEN VALLEY COLLEGE HAZARDOUS MATERIALS REPORT (SURVEY & INVENTORY) Dear Rachel: In an effort to assist the District in controlling costs and generating savings, Keenan & Associates recently conducted a Hazardous Materials Inventory & Survey for the District. The inventory is a valuable cost saving tool that: Reduces regulatory burden of inventorying and maintaining Material Safety Data Sheets Helps ensure proper quantities of materials used for custodial, science/art instruction and industrial purposes are maintained Minimizes waste disposal costs related to unused and outdated materials Assists in lowering quantities of stored materials and risks related with such storage like property damage, environmental impairment and health threats Ensures compliance and reduces the likelihood of visits and subsequent citations from regulatory agencies The Hazardous Materials Inventory and Survey includes: A Hazardous Materials Inventory (HMI) that provides the District with a detailed inventory of the hazardous materials observed at all applicable sites and locations throughout the District. In addition, the inventory is required for compliance with Hazardous Materials Business Plans. A Hazardous Materials Survey that addresses and identifies specific conditions regarding storage, labeling, compatibility, fire extinguishers, eyewash stations, etc., which were present at the time of the inventory. The survey will assist the District in complying with a number of Cal/OSHA regulations regarding chemicals in the workplace.