Endothelial Dysfunction During Graft-Versus-Host Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 20 *Lecture Powerpoint the Circulatory System: Blood Vessels and Circulation
Chapter 20 *Lecture PowerPoint The Circulatory System: Blood Vessels and Circulation *See separate FlexArt PowerPoint slides for all figures and tables preinserted into PowerPoint without notes. Copyright © The McGraw-Hill Companies, Inc. Permission required for reproduction or display. Introduction • The route taken by the blood after it leaves the heart was a point of much confusion for many centuries – Chinese emperor Huang Ti (2697–2597 BC) believed that blood flowed in a complete circuit around the body and back to the heart – Roman physician Galen (129–c. 199) thought blood flowed back and forth like air; the liver created blood out of nutrients and organs consumed it – English physician William Harvey (1578–1657) did experimentation on circulation in snakes; birth of experimental physiology – After microscope was invented, blood and capillaries were discovered by van Leeuwenhoek and Malpighi 20-2 General Anatomy of the Blood Vessels • Expected Learning Outcomes – Describe the structure of a blood vessel. – Describe the different types of arteries, capillaries, and veins. – Trace the general route usually taken by the blood from the heart and back again. – Describe some variations on this route. 20-3 General Anatomy of the Blood Vessels Copyright © The McGraw-Hill Companies, Inc. Permission required for reproduction or display. Capillaries Artery: Tunica interna Tunica media Tunica externa Nerve Vein Figure 20.1a (a) 1 mm © The McGraw-Hill Companies, Inc./Dennis Strete, photographer • Arteries carry blood away from heart • Veins -

Anatomy Review: Blood Vessel Structure & Function
Anatomy Review: Blood Vessel Structure & Function Graphics are used with permission of: Pearson Education Inc., publishing as Benjamin Cummings (http://www.aw-bc.com) Page 1. Introduction • The blood vessels of the body form a closed delivery system that begins and ends at the heart. Page 2. Goals • To describe the general structure of blood vessel walls. • To compare and contrast the types of blood vessels. • To relate the blood pressure in the various parts of the vascular system to differences in blood vessel structure. Page 3. General Structure of Blood Vessel Walls • All blood vessels, except the very smallest, have three distinct layers or tunics. The tunics surround the central blood-containing space - the lumen. 1. Tunica Intima (Tunica Interna) - The innermost tunic. It is in intimate contact with the blood in the lumen. It includes the endothelium that lines the lumen of all vessels, forming a smooth, friction reducing lining. 2. Tunica Media - The middle layer. Consists mostly of circularly-arranged smooth muscle cells and sheets of elastin. The muscle cells contract and relax, whereas the elastin allows vessels to stretch and recoil. 3. Tunica Adventitia (Tunica Externa) - The outermost layer. Composed of loosely woven collagen fibers that protect the blood vessel and anchor it to surrounding structures. Page 4. Comparison of Arteries, Capillaries, and Veins • Let's compare and contrast the three types of blood vessels: arteries, capillaries, and veins. • Label the artery, capillary and vein. Also label the layers of each. • Arteries are vessels that transport blood away from the heart. Because they are exposed to the highest pressures of any vessels, they have the thickest tunica media. -

Blood Vessels: Part A
Chapter 19 The Cardiovascular System: Blood Vessels: Part A Blood Vessels • Delivery system of dynamic structures that begins and ends at heart – Arteries: carry blood away from heart; oxygenated except for pulmonary circulation and umbilical vessels of fetus – Capillaries: contact tissue cells; directly serve cellular needs – Veins: carry blood toward heart Structure of Blood Vessel Walls • Lumen – Central blood-containing space • Three wall layers in arteries and veins – Tunica intima, tunica media, and tunica externa • Capillaries – Endothelium with sparse basal lamina Tunics • Tunica intima – Endothelium lines lumen of all vessels • Continuous with endocardium • Slick surface reduces friction – Subendothelial layer in vessels larger than 1 mm; connective tissue basement membrane Tunics • Tunica media – Smooth muscle and sheets of elastin – Sympathetic vasomotor nerve fibers control vasoconstriction and vasodilation of vessels • Influence blood flow and blood pressure Tunics • Tunica externa (tunica adventitia) – Collagen fibers protect and reinforce; anchor to surrounding structures – Contains nerve fibers, lymphatic vessels – Vasa vasorum of larger vessels nourishes external layer Blood Vessels • Vessels vary in length, diameter, wall thickness, tissue makeup • See figure 19.2 for interaction with lymphatic vessels Arterial System: Elastic Arteries • Large thick-walled arteries with elastin in all three tunics • Aorta and its major branches • Large lumen offers low resistance • Inactive in vasoconstriction • Act as pressure reservoirs—expand -

Vocabulario De Morfoloxía, Anatomía E Citoloxía Veterinaria
Vocabulario de Morfoloxía, anatomía e citoloxía veterinaria (galego-español-inglés) Servizo de Normalización Lingüística Universidade de Santiago de Compostela COLECCIÓN VOCABULARIOS TEMÁTICOS N.º 4 SERVIZO DE NORMALIZACIÓN LINGÜÍSTICA Vocabulario de Morfoloxía, anatomía e citoloxía veterinaria (galego-español-inglés) 2008 UNIVERSIDADE DE SANTIAGO DE COMPOSTELA VOCABULARIO de morfoloxía, anatomía e citoloxía veterinaria : (galego-español- inglés) / coordinador Xusto A. Rodríguez Río, Servizo de Normalización Lingüística ; autores Matilde Lombardero Fernández ... [et al.]. – Santiago de Compostela : Universidade de Santiago de Compostela, Servizo de Publicacións e Intercambio Científico, 2008. – 369 p. ; 21 cm. – (Vocabularios temáticos ; 4). - D.L. C 2458-2008. – ISBN 978-84-9887-018-3 1.Medicina �������������������������������������������������������������������������veterinaria-Diccionarios�������������������������������������������������. 2.Galego (Lingua)-Glosarios, vocabularios, etc. políglotas. I.Lombardero Fernández, Matilde. II.Rodríguez Rio, Xusto A. coord. III. Universidade de Santiago de Compostela. Servizo de Normalización Lingüística, coord. IV.Universidade de Santiago de Compostela. Servizo de Publicacións e Intercambio Científico, ed. V.Serie. 591.4(038)=699=60=20 Coordinador Xusto A. Rodríguez Río (Área de Terminoloxía. Servizo de Normalización Lingüística. Universidade de Santiago de Compostela) Autoras/res Matilde Lombardero Fernández (doutora en Veterinaria e profesora do Departamento de Anatomía e Produción Animal. -

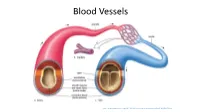
Blood Vessels
BLOOD VESSELS Blood vessels are how blood travels through the body. Whole blood is a fluid made up of red blood cells (erythrocytes), white blood cells (leukocytes), platelets (thrombocytes), and plasma. It supplies the body with oxygen. SUPERIOR AORTA (AORTIC ARCH) VEINS & VENA CAVA ARTERIES There are two basic types of blood vessels: veins and arteries. Veins carry blood back to the heart and arteries carry blood from the heart out to the rest of the body. Factoid! The smallest blood vessel is five micrometers wide. To put into perspective how small that is, a strand of hair is 17 micrometers wide! 2 BASIC (ARTERY) BLOOD VESSEL TUNICA EXTERNA TUNICA MEDIA (ELASTIC MEMBRANE) STRUCTURE TUNICA MEDIA (SMOOTH MUSCLE) Blood vessels have walls composed of TUNICA INTIMA three layers. (SUBENDOTHELIAL LAYER) The tunica externa is the outermost layer, primarily composed of stretchy collagen fibers. It also contains nerves. The tunica media is the middle layer. It contains smooth muscle and elastic fiber. TUNICA INTIMA (ELASTIC The tunica intima is the innermost layer. MEMBRANE) It contains endothelial cells, which TUNICA INTIMA manage substances passing in and out (ENDOTHELIUM) of the bloodstream. 3 VEINS Blood carries CO2 and waste into venules (super tiny veins). The venules empty into larger veins and these eventually empty into the heart. The walls of veins are not as thick as those of arteries. Some veins have flaps of tissue called valves in order to prevent backflow. Factoid! Valves are found mainly in veins of the limbs where gravity and blood pressure VALVE combine to make venous return more 4 difficult. -

Nomina Histologica Veterinaria, First Edition
NOMINA HISTOLOGICA VETERINARIA Submitted by the International Committee on Veterinary Histological Nomenclature (ICVHN) to the World Association of Veterinary Anatomists Published on the website of the World Association of Veterinary Anatomists www.wava-amav.org 2017 CONTENTS Introduction i Principles of term construction in N.H.V. iii Cytologia – Cytology 1 Textus epithelialis – Epithelial tissue 10 Textus connectivus – Connective tissue 13 Sanguis et Lympha – Blood and Lymph 17 Textus muscularis – Muscle tissue 19 Textus nervosus – Nerve tissue 20 Splanchnologia – Viscera 23 Systema digestorium – Digestive system 24 Systema respiratorium – Respiratory system 32 Systema urinarium – Urinary system 35 Organa genitalia masculina – Male genital system 38 Organa genitalia feminina – Female genital system 42 Systema endocrinum – Endocrine system 45 Systema cardiovasculare et lymphaticum [Angiologia] – Cardiovascular and lymphatic system 47 Systema nervosum – Nervous system 52 Receptores sensorii et Organa sensuum – Sensory receptors and Sense organs 58 Integumentum – Integument 64 INTRODUCTION The preparations leading to the publication of the present first edition of the Nomina Histologica Veterinaria has a long history spanning more than 50 years. Under the auspices of the World Association of Veterinary Anatomists (W.A.V.A.), the International Committee on Veterinary Anatomical Nomenclature (I.C.V.A.N.) appointed in Giessen, 1965, a Subcommittee on Histology and Embryology which started a working relation with the Subcommittee on Histology of the former International Anatomical Nomenclature Committee. In Mexico City, 1971, this Subcommittee presented a document entitled Nomina Histologica Veterinaria: A Working Draft as a basis for the continued work of the newly-appointed Subcommittee on Histological Nomenclature. This resulted in the editing of the Nomina Histologica Veterinaria: A Working Draft II (Toulouse, 1974), followed by preparations for publication of a Nomina Histologica Veterinaria. -

Blood Vessels
10/1/2010 Objectives Overview of the vessels. Human Anatomy & Define the different type of blood vessels. Physiology BI 233 --ArteriesArteries --VeinsVeins Course Intro --CapillariesCapillaries Identify the tissue layers of blood vessels. Distinguish the different forms of arteries, veins and capillaries. Homework Preparations Function of the Blood Vessels Cardiac Flow in HW section (diagram) Transport blood to and from the heart. Need colored pencils or marker Due Wed I. Arteries deliver oxygenated blood to tissues. HW #1 Artery Labeling II Veins carry deoxygenated blood to the Due in lab (Thursday) heart. III Capillaries are the site of gas exchange for internal respiration. Venous system Arterial system Tunica intima Large veins Heart (capacitance • Endothelium Valve • Subendothelial layer vessels) Elastic arteries Large (conducting Internal elastic lamina lymphatic vessels) Tunica media vessels (smooth muscle and Lymph elastic fibers) node Muscular arteries (distributing External elastic lamina Lymphatic system vessels) Tunica externa Small veins (collagen fibers) Arteriovenous (capacitance anastomosis vessels) Lymphatic Sinusoid capillary Lumen Arterioles Lumen Capillary Vein (resistance vessels) Artery network Postcapillary Terminal arteriole Basement membrane venule Metarteriole Endothelial cells Thoroughfare Capillaries Precapillary sphincter channel (exchange vessels) (b) Capillary Copyright © 2010 Pearson Education, Inc. Copyright © 2010 Pearson Education, Inc. 1 10/1/2010 Tissue Layers of Vessels Tissue Layers of Vessels -

Morphometric Analysis of Thoracic Aorta in Slc39a13/Zip13-KO Mice
Cell and Tissue Research (2019) 376:137–141 https://doi.org/10.1007/s00441-018-2977-9 SHORT COMMUNICATION Morphometric analysis of thoracic aorta in Slc39a13/Zip13-KO mice Takuya Hirose1 & Takamasa Shimazaki1 & Naoki Takahashi1 & Toshiyuki Fukada2 & Takafumi Watanabe1,3 & Prasarn Tangkawattana 1,4 & Kazushige Takehana1 Received: 18 July 2018 /Accepted: 13 October 2018 /Published online: 4 January 2019 # Springer-Verlag GmbH Germany, part of Springer Nature 2019 Abstract Ehlers–Danlos syndrome (EDS) is a collection of inheritable diseases involving the musculoskeletal, integumentary and visual systems. Spondylodysplastic EDS-ZIP13 (spEDS-ZIP13: OMIM 612350) was recently defined as a new form of EDS. Although vasculitis has been found in many spEDS-ZIP13 patients, vascular pathology has not been included as a pathognomonic lesion of this type of EDS. We investigate the morphometry of the thoracic aorta in wild-type and Zip13-knockout (Zip13-KO) mice. Our assessment found abnormalities in the number and morphology of elastic and cellular components in the aortic wall, especially the tunica media, of Zip13-KO mice, indicating aortic fragility. Accordingly, our major findings (vascular smooth muscle cells with small nuclei, small percentage of elastic membrane area per tunica media, many large elastic flaps) should be considered vulnerable characteristics indicating fragility of the aorta in patients with spEDS-ZIP13. Keywords Aorta . Elasticity . Elastic membrane . Morphometry . Slc39a13/Zip13-KO mouse Introduction (Fukada et al. 2008). ZIP13, a member of the SLC39/ZIP family, is an intercellular zinc transporter located in the Ehlers–Danlos syndrome (EDS) is a collection of hereditary Golgi complex of osteoblasts, chondrocytes, pulpal cells diseases that manifest as skin hyperextension, joint hypermo- and fibroblasts (Fukada et al. -

The Circulatory System
The Circulatory System Lec.10 Histology 1 The Circulatory System includes both 1. blood vascular system 2. lymphatic vascular system 2 blood vascular system 1. The heart, an organ whose function is to pump the blood. 2. The arteries, a series of efferent vessels that become smaller as they branch, and whose function is to carry the blood, with its nutrients and oxygen, to the tissues. 3. The capillaries, the smallest blood vessels, constituting a complex network of thin tubules that branch profusely in almost every organ and through whose walls the interchange between blood and tissues takes place. 4. The veins, which result from the convergence of capillaries into a system of larger channels that continue enlarging as they approach the heart, toward which they convey the blood to be pumped again. 3 The lymphatic vascular system • The lymphatic vascular system, begins with the lymphatic capillaries, which are closed-ended tubules that merge to form vessels of steadily increasing size; these vessels terminate in the blood vascular system emptying into the large veins near the heart. • One of the functions of the lymphatic system is to return the fluid of the tissue spaces to the blood. • The internal surface of all components of the blood and lymphatic systems is lined by a single layer of a squamous epithelium, called endothelium. 4 Heart • The heart is a muscular organ that contracts rhythmically, pumping the blood through the circulatory system . • The right and left ventricles pump blood to the lungs and the rest of the body respectively; right and left atria receive blood from the body and the pulmonary veins respectively. -

Vegfr3 and Notch Signaling in Angiogenesis
VEGFR3 AND NOTCH SIGNALING IN ANGIOGENESIS Georgia Zarkada, M.D. ACADEMIC DISSERTATION Wihuri Research Institute & Research Programs Unit Translational Cancer Biology Faculty of Medicine & Doctoral Program in Biomedicine University of Helsinki Finland To be publicly discussed, with the permission of the Faculty of Medicine, University of Helsinki, in Biomedicum Helsinki 1, Lecture Hall 2, Haartmaninkatu 8, on the 8th of August 2014, at 12 o’ clock noon. Helsinki 2014 VEGFR3 and Notch signaling in angiogenesis ! Supervised by Kari Alitalo M.D., Ph.D. Research Professor of the Finnish Academy of Sciences Wihuri Research Institute Translational Cancer Biology University of Helsinki Finland and Tuomas Tammela, M.D., Ph.D. Adjunct Professor Koch Institute for Integrative Cancer Research Massachusetts Institute of Technology Cambridge, MA USA Reviewed by Juha Partanen, Ph.D. Professor Department of Biosciences, Division of Genetics University of Helsinki Finland and Mariona Graupera, Ph.D. Bellvitge Biomedical Research Institute Catalan Institute of Oncology University of Barcelona Spain Opponent Christiana Ruhrberg, Ph.D. Professor UCL Institute of Ophthalmology University College London London UK ISBN 978-952-10-9997-7 ISSN 2342-3161 Hansaprint 2014 ! 2! VEGFR3 and Notch signaling in angiogenesis ! To my family ! 3! VEGFR3 and Notch signaling in angiogenesis ! TABLE OF CONTENTS ABBREVIATIONS………………………………………………………………………... 6 LIST OF ORIGINAL PUBLICATIONS………………………………………..……... 8 ABSTRACT…………………………………………………………………………..…... 9 INTRODUCTION………………………………………………………………..……… 11 REVIEW OF THE LITERATURE………………………………………………..…….. 12 1. ANATOMY AND FUNCTION OF THE VASCULAR SYSTEMS………..……. 12 1.1 The blood vascular system…………………………………………………..……… 12 1.1.1 Blood vessels structure and physiology……………………………….………….. 12 1.1.1.1 Regulation of vascular permeability……………………………..………….. 12 1.1.1.2 Regulation of flow…………………………………………………..………. -

Blood Vessels
Elastic artery Venules. Muscular (distributing) Small veins (medium-sized) artery Medium- Arterioles sized veins Large veins General Structure of Blood Vessels - The wall of blood vessel is formed of three concentric layers: Tunica intima (interna) Tunica media Tunica adventitia (externa) Tunica Intima Single layer of flattened Subendothelial layer made Beneath the subendothelial endothelial cells (resting on up of loose connective layer is an internal elastic the basal lamina) lining the tissue. May have few lamina, composed of elastin lumen of the vessel longitudinally arranged (fenestrated elastic sheet), smooth muscle fibers separating the tunica intima from the tunica media Tunica Media Composed of smooth Large muscular arteries have external elastic lamina, muscles, some elastic fibers, separating the tunica media from the tunica adventitia. type III collagen (reticular Capillaries and postcapillary venules do not have a tunica fibers) and type I collagen. media, however, pericytes replace the tunica media. Tunica Adventitia - Composed of connective . Vasa vasorum: “Outermost layer” tissue containing types I & III NN..BB. collagen, fibroblasts and are small arterioles in tunica adventitia longitudinal elastic fibers and the outer part of tunica media. - Blends into the surrounding They are more prevalent in the connective tissue. walls of veins than arteries – why? Venous blood contains less oxygen and nutrients than arterial blood. ELASTIC ARTERIES T. Adventitia T. Intima T. Media Much thinner than T.M Fenestrated elastic loose C.T *Endothelium. membranes (sheets) (lamellae) In between, there are: 1. Smooth muscle cells: Subendothelial C.T. * less abundant & secrete Contains vasa all other components in T.M. vasorum → send 2. Collagen fibers (type I branches to the collagen). -
Circulation-Post
Blood Vessels http://www.majordifferences.com/2013/02/difference-between-artery-and-vein.html#.WiSD2VVKt0w ateries veins • Carry blood away from the • Carry blood toward the heart heart • Elastic arteries resist • Low pressure pressure and even it out • Thin walls (partially) • Large lumen • Muscular arteries maintain • Valves to prevent backflow & control pressure and promote flow of blood • Arterioles control blood in only one direction flow into capillaries capillaries – only endothelium http://anatomybody101.com/the-heart-is-made-of-what- muscle/the-heart-is-made-of-what-muscle-blood-vessels- tunica-intima-endothelium-subendothelial-layer-internal-or- external-elastic-membrane-tunica-media-externa-basement- membrane-artery-vein-capillary-bed/ muscular artery – vein – nerve elastin stain http://zoomify.lumc.edu/histonew/cardiovascular/dms108/popup.html Diagram of the tunics of an arterial wall (above): On the left, the drawing illustrates a typical stain and the tunica media smooth muscle are apparent; on the right, the same arterial wall has been stained with a special stain for elastin and you can see the elastin fibers running between the smooth muscle cells of the tunica media as well as through the loose connective tissue of the tunica externa. http://www.apsubiology.org/anatomy/2020/2020_Exam_Reviews/Exa m_1/CH19_Vessel_Wall_Histology.htm Elastic artery – stained for elastin https://casweb.ou.edu/p bell/Histology/Caption s/Cardiovascular/46.ela stic.art.10x.html Capillaries come in 3 varieties – most are “continuous” though http://ib.bioninja.com.au/standard-level/topic-6-human-physiology/62-the-blood-system/capillaries.html