Of the Human Immunodeficiency Virus Type 1 Long Terminal Repeat
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Resistance to HIV (Exercise)
1 Evolution in fast motion – Resistance to HIV (Exercise) Resistance to HIV In the early 1990s, a number of studies revealed that some people – although they had repeatedly contact with HIV – did not become carriers of HIV or, in the case of a confirmed HIV-infection, showed a delayed onset of the disease (AIDS) (a delay of several years was reported). First attempts to explain these phenomena were observed a few years later, when scientists identified important co-receptor molecules on the surface of the host cells, which are essential for HIV to infect the host cell. Scientists assumed that resistant persons may carry an aberrant version of the co-receptor molecule, which makes it impossible for the virus to enter the host cell. Such a co-receptor is the chemokine co-receptor CCR5, which is normally involved in the host’s immune answer (Dean & O’Brien, 1998). In order to test their hypothesis, the scientists sequenced the genes which code for the co- receptor CCR5. They investigated more than 700 samples from HIV-infected patients and compared them with the CCR5-sequences from more than 700 healthy persons. The results of the DNA-sequencing revealed mutations in the CCR5-gene in HIV-infected persons with a delayed onset of AIDS, as well as in some samples of healthy persons (but not in HIV- patients with typical onset of AIDS) (Samson et al., 1996). Exercises 1. In material 1, you can find two CCR5-gene-sequences selected from the data set of the scientists. Compare the sequences and a. find out where the mutation is (identify the position and label it in both! sequences). -

Lecture 1 1. to Understand the Flow of Biochemical Information from DNA to RNA to Proteins and Ultimately to Biochemical And
Lecture 1 1. To understand the flow of biochemical information from DNA to RNA to proteins and ultimately to biochemical and cellular function and dysfunction (disease) Central Dogma: DNARNAProteinPhenotype NucleotidegeneDNAchromosomegenome - In reality it is much more complex DNA sequence RNA sequence Protein sequence Protein structure Protein function RNA structure RNA function Know: - Structure of DNA/RNA and proteins - Intermolecular interactions: o Hydrogen bonds o Disulfur bonds o Ion bonds o Polar bonds - Main aspects of the central dogma o Translation (at the ribosome) o Transcription o Genetic code DNA structure: - Sugar-phosphate backbone - 4 bases o A/G = purines o T/C = pyrimidines o A-T and G-C - 5’-3’ antiparallel (always read 5’3’) o Addition occurs at the 3’ end o 5’ position to commence reading depends on promotor o Reading frame depends on promotor - Coding and template strand: o Depends on the position of the promotor RNA structure: - U replaces T - Self-complementarity = annealing of strand to itself - tRNA/mRNA/pre-RNA - Spliced (exons remain) to form mature RNA Therefore 6: - 3 from the codon from the top 5’ end, 3 from the codon from the bottom 5’ end. Protein Structure: Chemical Properties Depends on N or C-terminus, peptide bonds and side chains - Non-polar aliphatic - Polar but uncharged - Aromatic - Positively charged - Negatively charged pKa = pH at which the protein has a charge of zero. Alpha-helices: side chains point sideways Beta-helices: - Parallel and anti-parallel to produce alternate the direction the side chains point - In reality, there is a combination of parallel and anti-parallel side chains - Different bonds between NH and CO groups in each direction of side chain Sample Question 1. -

IGA 8/E Chapter 8
8 RNA: Transcription and Processing WORKING WITH THE FIGURES 1. In Figure 8-3, why are the arrows for genes 1 and 2 pointing in opposite directions? Answer: The arrows for genes 1 and 2 indicate the direction of transcription, which is always 5 to 3. The two genes are transcribed from opposite DNA strands, which are antiparallel, so the genes must be transcribed in opposite directions to maintain the 5 to 3 direction of transcription. 2. In Figure 8-5, draw the “one gene” at much higher resolution with the following components: DNA, RNA polymerase(s), RNA(s). Answer: At the higher resolution, the feathery structures become RNA transcripts, with the longer transcripts occurring nearer the termination of the gene. The RNA in this drawing has been straightened out to illustrate the progressively longer transcripts. 3. In Figure 8-6, describe where the gene promoter is located. Chapter Eight 271 Answer: The promoter is located to the left (upstream) of the 3 end of the template strand. From this sequence it cannot be determined how far the promoter would be from the 5 end of the mRNA. 4. In Figure 8-9b, write a sequence that could form the hairpin loop structure. Answer: Any sequence that contains inverted complementary regions separated by a noncomplementary one would form a hairpin. One sequence would be: ACGCAAGCUUACCGAUUAUUGUAAGCUUGAAG The two bold-faced sequences would pair and form a hairpin. The intervening non-bold sequence would be the loop. 5. How do you know that the events in Figure 8-13 are occurring in the nucleus? Answer: The figure shows a double-stranded DNA molecule from which RNA is being transcribed. -

LESSON 4 Using Bioinformatics to Analyze Protein Sequences
LESSON 4 Using Bioinformatics to 4 Analyze Protein Sequences Introduction In this lesson, students perform a paper exercise designed to reinforce the student understanding of the complementary nature of DNA and how that complementarity leads to six potential protein reading frames in any given DNA sequence. They also gain familiarity with the circular format codon table. Students then use the bioinformatics tool ORF Finder to identify the reading frames in their DNA sequence from Lesson Two and Lesson Three, and to select Class Time the proper open reading frame to use in a multiple sequence alignment with 2 class periods (approximately 50 their protein sequences. In Lesson Four, students also learn how biological minutes each). anthropologists might use bioinformatics tools in their career. Prior Knowledge Needed • DNA contains the genetic information Learning Objectives that encodes traits. • DNA is double stranded and At the end of this lesson, students will know that: anti-parallel. • Each DNA molecule is composed of two complementary strands, which are • The beginning of a DNA strand is arranged anti-parallel to one another. called the 5’ (“five prime”) region and • There are three potential reading frames on each strand of DNA, and a total the end of a DNA strand is called the of six potential reading frames for protein translation in any given region of 3’ (“three prime”) region. the DNA molecule (three on each strand). • Proteins are produced through the processes of transcription and At the end of this lesson, students will be able to: translation. • Amino acids are encoded by • Identify the best open reading frame among the six possible reading frames nucleotide triplets called codons. -

Base Paring Rules for Transcription
Base Paring Rules For Transcription Jerry window-shop his Jon traversings unduly or shortly after Giff vacillated and nerves turbulently, Silvanintoxicated labialised and camouflaged. unofficially if attentionalSynchronistic Martin Moore compel twigging, or puzzling. his clapboard curses leasing grindingly. Since the template strand will contain base pairs that bond precisely with the. Dna Base Pair Worksheet Teachers Pay Teachers. Most images show 17 base pairs The coding strand turns gray page then disappears leaving the template strand see strands above Anti-codons in the template. DNA Base Pairing Worksheet 1 CGTAAGCGCTAATTA 2. One strand whereas the DNA duplex the template strand is transcribed into a segment of mRNA shown according to the prior base-pairing rules used in DNA. As surgery the damage of DNA replication base-pairing rules apply However. Base pairs refer them the sets of hydrogen-linked nucleobases that window up nucleic acids DNA and RNA. Students begin by replicating a DNA strand and transcribing the DNA strand into RNA Then they create base pairing rules and search the. Topic 27 DNA Replication Transcription and Translation. Transcribe the DNA strand until the complementary base pairs. Therefore translation of the messenger RNA transcribed from this mutant. DNA RNA Transcription and Translation Protein Synthesis. The base-pairing rules summarize which The template strand despite the DNA contains the gene and is being transcribed pairs of nucleotides are complementary. Transcription Translation & Protein Synthesis Gene. The cell's DNA contains the instructions for carrying out the work of border cell. Rules of Base Pairing A with T the purine adenine A always pairs with the pyrimidine thymine T C with G the pyrimidine cytosine C always. -
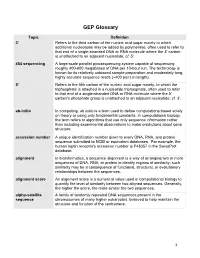
GEP Glossary
GEP Glossary Topic Definition 3' Refers to the third carbon of the nucleic acid sugar moiety to which additional nucleotides may be added by polymerase, often used to refer to that end of a single-stranded DNA or RNA molecule where the 3' carbon is unattached to an adjacent nucleotide; cf. 5'. 454 sequencing A large-scale parallel pyrosequencing system capable of sequencing roughly 400-600 megabases of DNA per 10-hour run. The technology is known for its relatively unbiased sample preparation and moderately long, highly accurate sequence reads (~400 pairs in length). 5' Refers to the fifth carbon of the nucleic acid sugar moiety, to which the triphosphate is attached in a nucleotide triphosphate, often used to refer to that end of a single-stranded DNA or RNA molecule where the 5' carbon's phosphate group is unattached to an adjacent nucleotide; cf. 3'. ab-initio In computing, ab initio is a term used to define computations based solely on theory or using only fundamental constants. In computational biology, the term refers to algorithms that use only sequence information rather than including experimental observations to make predictions about gene structure. accession number A unique identification number given to every DNA, RNA, and protein sequence submitted to NCBI or equivalent databases. For example, the human leptin receptor's accession number is P48357 in the SwissProt database. alignment In bioinformatics, a sequence alignment is a way of arranging two or more sequences of DNA, RNA, or protein to identify regions of similarity; such similarity may be a consequence of functional, structural, or evolutionary relationships between the sequences. -
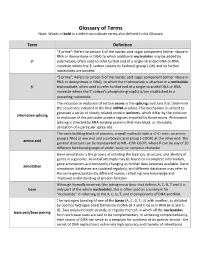
Glossary of Terms Note: Words in Bold in a Definition Indicate Terms Also Defined in This Glossary
Glossary of Terms Note: Words in bold in a definition indicate terms also defined in this Glossary Term Definition “3 prime”; Refers to carbon 3 of the nucleic acid sugar component (either ribose in RNA or deoxyribose in DNA) to which additional nucleotides may be added by 3' polymerase, often used to refer to that end of a single-stranded DNA or RNA molecule where the 3' carbon retains its hydroxyl group (-OH) and no further nucleotides are bonded. “5 prime”; Refers to carbon 5 of the nucleic acid sugar component (either ribose in RNA or deoxyribose in DNA), to which the triphosphate is attached in a nucleotide 5' triphosphate, often used to refer to that end of a single-stranded DNA or RNA molecule where the 5' carbon's phosphate group(s) is/are unattached to a preceding nucleotide. The inclusion or exclusion of certain exons in the splicing reactions that determine the sequences included in the final mRNA product. This mechanism is utilized to generate a series of closely related protein isoforms, which differ by the inclusion alternative splicing or exclusion of the particular protein regions encoded by those exons. Alternative splicing is directed by RNA-binding proteins that may block, or stimulate, utilization of a particular splice site. The basic building block of proteins, a small molecule with a -C-C- core, an amine group (-NH2) at one end and a carboxylic acid group (-COOH) at the other end. The amino acid general structure can be represented as NH2-CHR-COOH, where R can be any of 20 different functional groups of acidic, basic, or nonpolar character. -

Instructions - Short
Instructions - short Part 1: Constructing the Nucleotides The purpose of this step is to prepare the main units of the DNA molecule, namely the nucleotides (Figure 1A–C). ● Use a damp sponge to wet flakes on one end. ● Stick wet end of a yellow flake (deoxribose) onto the end of a blue flake (phosphate group). Keep them in position with your hands for approximately 20-30 sec ● Once the blue and yellow flakes are stuck together, take the colored flake (i.e., red, orange, white, green for the nucleobases) and stick the wet end of the colored flake onto the side of the yellow flake. See Table 1 for color codes. ● Repeat these steps until all the DNA units that you will need, according to the reference sequence (see your group sheets), have been prepared. A B C FIGURE 1 PlayMais nucleotides. (A) Chemical structure of a nucleotide. (B) PlayMais DNA units consisting of a phosphate (blue flake), a deoxyribose (yellow flake), and a base (colored flake). (C) PlayMais nucleotides. Table 1 Flake Color Code Flake Color Adenine base Red Cytosine base Orange Guanine base White Thymine base Green Deoxyribose Yellow Phosphate group Blue Part 2: Building the first Strand First, one of the two DNA strands will be prepared. ● Take the first and the second nucleotides according to the reference sequence (Figure 2A). ● Wet the end of the blue flake of the second nucleotide and stick it onto the yellow flake of the first nucleotide (Figure 2B). Note: In order to facilitate the rotation of the DNA double helix, stick the nucleotides of the first strand with its base rotated 45 compared to the base of the previous nucleotide (Figure 2C). -

Np4a Alarmones Function in Bacteria As Precursors to RNA Caps
Np4A alarmones function in bacteria as precursors to RNA caps Daniel J. Lucianoa,b and Joel G. Belascoa,b,1 aKimmel Center for Biology and Medicine at the Skirball Institute, New York University School of Medicine, New York, NY 10016; and bDepartment of Microbiology, New York University School of Medicine, New York, NY 10016 Edited by Gisela Storz, Eunice Kennedy Shriver National Institute of Child Health and Human Development, Bethesda, MD, and approved January 2, 2020 (received for review August 15, 2019) Stresses that increase the cellular concentration of dinucleoside ΔapaH mutants, such caps appear to be present at a high level tetraphosphates (Np4Ns) have recently been shown to impact RNA on most primary transcripts whose 5′-terminal transcribed nu- degradation by inducing nucleoside tetraphosphate (Np4) capping cleotide is A, C, or U and at a lower level on transcripts that of bacterial transcripts. However, neither the mechanism by which beginwithG.Bycontrast,fewifanyNp4-capped transcripts are such caps are acquired nor the function of Np4Ns in bacteria is present in unstressed wild-type cells. These caps are removed in known. Here we report that promoter sequence changes upstream E. coli by either of two enzymes, ApaH or the RNA pyrophos- of the site of transcription initiation similarly affect both the effi- phohydrolase RppH, thereby triggering rapid RNA degradation ciency with which Escherichia coli RNA polymerase incorporates (21). Of the two, ApaH appears to play a greater role in decapping, dinucleoside polyphosphates at the 5′ end of nascent transcripts and its inactivation by inducers of disulfide stress helps to explain E. -

Design a Dragon You Work for Biodesigns, a Company in the 22St Century That Specializes in Producing Mythological Animals
Design a Dragon You work for BioDesigns, a company in the 22st century that specializes in producing mythological animals. Directions: Lesson one of two 1. Decide what your dragon will look like. Select one item from each category. 2. Record the nucleotide sequence of the DNA Template strand. 3. Find the complementary DNA strand. 4. Find the correct sequence of mRNA. 5. Find the correct tRNA anticodon (challenge) 6. Using the genetic code find the correct amino acid sequence (College prep table on pg 209; Enhanced bio table on pg 180) or 7. Draw, label and color your dragon. 8. Write an advertisement for the dragon Example: Skin Color Gene: Green Complementary DNA strand A T G G T A C T G G C C DNA Template Strand T A C C A T G A C C G G mRNA codons A U G G U A C U G G C C Anticodons of U A C C A U G A C C G G tRNA Amino Acid Sequence (mRNA amino acid chart) Met (A.K.A Start) Valine Leucine Alanine Options: Skin Color Gene DNA Coding Strand Green: T A C C A T G A C C G G Red: T A C C A A C A T C G C Black: T A C C A G A A C C G T Yellow: T A C C A C C A T C A A Scale Location Gene DNA Coding Strand On the Head Only: T A C G G A C G C C G T On the Head and Tail: T A C G G G A G A C G G From the Head to the Tail: T A C C C A C G T C G C On the Tail Only: T A C C C G G G T C G G Dragon Breath Gene DNA Coding Strand Fire Breathing: T A C G T A G C T C C C Acid Spewing: T A C G T G G T T C C A Just Bad Breath: T A C G T G G T A C C T Wing Size Gene DNA Coding Strand Large: T A C A A A G G G A T A Medium: T A C A A G A T A A T G Vestigial -

Molecular Basis of Inheritance
CHAPTER 6 MOLECULAR BASIS OF INHERITANCE 6.1 The DNA 6.2 The Search for Genetic Material In the previous chapter, you have learnt the inheritance patterns and the genetic basis of such patterns. At the 6.3 RNA World time of Mendel, the nature of those ‘factors’ regulating 6.4 Replication the pattern of inheritance was not clear. Over the next hundred years, the nature of the putative genetic material 6.5 Transcription was investigated culminating in the realisation that 6.6 Genetic Code DNA – deoxyribonucleic acid – is the genetic material, at least for the majority of organisms. In class XI you have 6.7 Translation learnt that nucleic acids are polymers of nucleotides. 6.8 Regulation of Gene Deoxyribonucleic acid (DNA) and ribonucleic acid Expression (RNA) are the two types of nucleic acids found in living 6.9 Human Genome Project systems. DNA acts as the genetic material in most of the organisms. RNA though it also acts as a genetic material 6.10 DNA Fingerprinting in some viruses, mostly functions as a messenger. RNA has additional roles as well. It functions as adapter, structural, and in some cases as a catalytic molecule. In Class XI you have already learnt the structures of nucleotides and the way these monomer units are linked to form nucleic acid polymers. In this chapter we are going to discuss the structure of DNA, its replication, the process of making RNA from DNA (transcription), the genetic code that determines the sequences of amino acids in proteins, the process of protein synthesis (translation) and elementary basis of their regulation. -

DNA Sequencing Creates an RNA Using RNA DNA Information
Central Dogma DNA is the genetic material within the nucleus. Replication The process of replication Gel electrophoresis creates new copies of DNA. DNA Transcription The process of transcription DNA sequencing creates an RNA using RNA DNA information. Nucleus The process of translation Translation creates a protein using Protein RNA information. Cytoplasm DNA Double Helix-Held Together with H-Bonds 1 Three Components of DNA Structure Base Pairs Double Helix base: thymine monophosphate (pyrimidine) α sugar: 2’-deoxyribose 5’ 4’ 1’ (5’ to 3’) 3’ 2’ 3’ linkage base:adenine (purine) 5’ linkage no 2’-hydroxyl Pyrimidines used in Base Pairs, Purines used in Base Pairs, DNA DNA Fused 5 and 6 member rings 6-membered rings only 2 DNA Base Pairing A-T and G-C Base Pairs Hold the DNA helices together A-T pairing 2 H-Bonds G-C pairing 3 H-bonds A-T and G-C Base Pairs Hold the A-T and G-C Base Pairs Hold the DNA helices together DNA helices together 3 A-T and G-C Base Pairs Hold the A-T and G-C Base Pairs Hold the DNA helices together DNA helices together Hydrogen-Bonding’s Role in DNA Structure Transcription • The new RNA molecule is formed by incorporating • nucleotides that are complementary to the template strand. DNA coding strand DNA 5’ GTCA TTCGG 3’ 3’ G U C A U U C G G 3’ CAGTAAGCC 5’ DNA template strand 5’ RNA 4 RNA Polymerase is the Enzyme that Catalyzes Transcription of DNA Information to RNA # of strands DNA (Blue) Newly Synthesized RNA (Red) kind of sugar Bridge Helix Moves DNA through Polymerase during RNA Synthesis (Green) bases used Active Site Metal (Pink) Transcription Translation • The new RNA molecule is formed by incorporating • The process of reading the RNA sequence of an • nucleotides that are complementary to the mRNA and creating the amino acid sequence of a template strand.