THE REGULATION of HEPATIC CHOLINE TRANSPORT By
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Concise Guide to Pharmacology 2019/20
Edinburgh Research Explorer THE CONCISE GUIDE TO PHARMACOLOGY 2019/20 Citation for published version: Cgtp Collaborators 2019, 'THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters', British Journal of Pharmacology, vol. 176 Suppl 1, pp. S397-S493. https://doi.org/10.1111/bph.14753 Digital Object Identifier (DOI): 10.1111/bph.14753 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: British Journal of Pharmacology General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 28. Sep. 2021 S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Transporters. British Journal of Pharmacology (2019) 176, S397–S493 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters Stephen PH Alexander1 , Eamonn Kelly2, Alistair Mathie3 ,JohnAPeters4 , Emma L Veale3 , Jane F Armstrong5 , Elena Faccenda5 ,SimonDHarding5 ,AdamJPawson5 , Joanna L -

Interplay Between Metformin and Serotonin Transport in the Gastrointestinal Tract: a Novel Mechanism for the Intestinal Absorption and Adverse Effects of Metformin
INTERPLAY BETWEEN METFORMIN AND SEROTONIN TRANSPORT IN THE GASTROINTESTINAL TRACT: A NOVEL MECHANISM FOR THE INTESTINAL ABSORPTION AND ADVERSE EFFECTS OF METFORMIN Tianxiang Han A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Eshelman School of Pharmacy. Chapel Hill 2013 Approved By: Dhiren R. Thakker, Ph.D. Michael Jay, Ph.D. Kim L. R. Brouwer, Pharm.D., Ph.D. Joseph W. Polli, Ph.D. Xiao Xiao, Ph.D. © 2013 Tianxiang Han ALL RIGHTS RESERVED ii ABSTRACT TIANXIANG HAN: Interplay between Metformin and Serotonin Transport in the Gastrointestinal Tract: A Novel Mechanism for the Intestinal Absorption and Adverse Effects of Metformin (Under the direction of Dhiren R. Thakker, Ph.D.) Metformin is a widely prescribed drug for Type II diabetes mellitus. Previous studies have shown that this highly hydrophilic and charged compound traverses predominantly paracellularly across the Caco-2 cell monolayer, a well-established model for human intestinal epithelium. However, oral bioavailability of metformin is significantly higher than that of the paracellular probe, mannitol (~60% vs ~16%). Based on these observations, the Thakker laboratory proposed a “sponge” hypothesis (Proctor et al., 2008) which states that the functional synergy between apical (AP) transporters and paracellular transport enhances the intestinal absorption of metformin. This dissertation work aims to identify AP uptake transporters of metformin, determine their polarized localization, and elucidate their roles in the intestinal absorption and adverse effects of metformin. Chemical inhibition and transporter-knockdown studies revealed that four transporters, namely, organic cation transporter 1 (OCT1), plasma membrane monoamine transporter (PMAT), serotonin reuptake transporter (SERT) and choline high-affinity transporter (CHT) contribute to AP uptake of metformin in Caco-2 cells. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Prokaryotic Solute/Sodium Symporters: Versatile Functions and Mechanisms of a Transporter Family †
Review Prokaryotic Solute/Sodium Symporters: Versatile Functions and Mechanisms of a Transporter Family † Tania Henriquez, Larissa Wirtz, Dan Su and Heinrich Jung * Microbiology, Department Biology 1, Ludwig Maximilians University Munich, D-82152 Martinsried, Germany; [email protected] (T.H.); [email protected] (L.W.); [email protected] (D.S.) * Correspondence: [email protected]; Tel.: +49-89-218074630 † This article is dedicated to the memory of Ron Kaback, an exceptional scientist and great mentor. Abstract: The solute/sodium symporter family (SSS family; TC 2.A.21; SLC5) consists of integral membrane proteins that use an existing sodium gradient to drive the uphill transport of various solutes, such as sugars, amino acids, vitamins, or ions across the membrane. This large family has representatives in all three kingdoms of life. The human sodium/iodide symporter (NIS) and the sodium/glucose transporter (SGLT1) are involved in diseases such as iodide transport defect or glu- cose-galactose malabsorption. Moreover, the bacterial sodium/proline symporter PutP and the so- dium/sialic acid symporter SiaT play important roles in bacteria–host interactions. This review fo- cuses on the physiological significance and structural and functional features of prokaryotic mem- bers of the SSS family. Special emphasis will be given to the roles and properties of proteins con- taining an SSS family domain fused to domains typically found in bacterial sensor kinases. Citation: Henriquez, T.; Wirtz, L.; Keywords: secondary transport; solute/sodium symport; SLC5; PutP; signal transduction; bacterial Su, D.; Jung, H. Prokaryotic two-component systems; bacterial sensor kinase Solute/Sodium Symporters: Versatile Functions and Mechanisms of a Transporter Family. -

Molecular Characterization of Neurotransmitter Transporters
Molecular Characterization of Neurotransmitter Transporters Saad Shafqat, Maria Velaz-Faircloth, Ana Guadaiio-Ferraz, and Robert T. Fremeau, Jr. Downloaded from https://academic.oup.com/mend/article/7/12/1517/2714704 by guest on 06 October 2020 Departments of Pharmacology and Neurobiology Duke University Medical Center Durham, North Carolina 27710 INTRODUCTION ogical processes. For example, blockade and/or rever- sal of the high affinity plasma membrane L-glutamate transporter during ischemia or anoxia elevates the ex- Rapid chemical signaling between neurons and target tracellular concentration of L-glutamate to neurotoxic cells is dependent upon the precise control of the levels resulting in nerve cell damage (7). Conversely, concentration and duration of neurotransmitters in syn- specific GABA uptake inhibitors are being developed as aptic spaces. After transmitter release from activated potential anticonvulsant and antianxiety agents (8). The nerve terminals, the principal mechanism involved in the ability of synaptic transporters to accumulate certain rapid clearance from the synapse of the biogenic amine neurotransmitter-like toxins, including N-methyW and amino acid neurotransmitters is active transport of phenylpyridine (MPP+), 6-hydroxydopamine, and 5,6- the transmitter back into presynaptic nerve terminals or dihydroxytryptamine, suggests a role for these active glial surrounding cells by one of a large number of transport proteins in the selective vulnerability of neu- specific, pharmacologically distinguishable membrane rons -
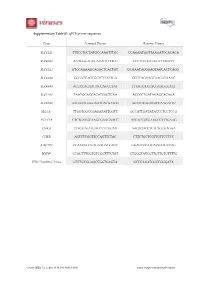
Gene Forward Primer Reverse Primer
Supplementary Table S1. qPCR primer sequences. Gene Forward Primer Reverse Primer SLC44A1 TTTCCTGCTATGCCAAGTTTGC CCAGAATGGTTAAGATCCACACA SLC44A2 AAAGGGAGGGAGAGTTTTGC CCCTTGGGTGGGTTTAGTTT SLC44A3 GTCCAAAAGCAGACTCACTGT GCAAATAGGGAGTAGCACTCAGG SLC44A4 GGGATCAGCGGTCTTATTGA GCGCAGAAGCAAGATAAAC SLC44A5 ACCCCAGAAGAGCAGCCTAT TTTAGCAACACGGAGGGACT SLC22A1 TAATGGACCACATCGCTCAA ACCCCTGATAGAGCACAGA SLC22A2 AAGAATGGGGAATCACAATGG AGATGTGGACGCCAAGATTC SLC5A7 TTGGTGGCCGAGATATTGGTT GCCATTGATATACCCTCCTCCG PCYT1A CTCTGATGCAAGCGAAGAACC ATCACCGTGAAGCCTTTGAAG CHKΑ CTTGGTGATGAGCCTCGGAA AAGTGACCTCTCTGCGAGAA CHKB AGTCTCGGTTCCAGTTCTAC CTTCTGCTCGTTGTTCCTCC β ACTIN CCAACCGCGAGAAGATGACC GGAGTCCATCACGATGCCAG HSP90 CCAGTTTGGTGTCGGTTTCTAT CTGGGTATCGTTGTTGTGTTTTG JFH1 Hepatitis C Virus GTCTGCGGAACCGGTGAGTA GCCCAAATGGCCGGGATA Viruses 2020, 12, x; doi: FOR PEER REVIEW www.mdpi.com/journal/viruses Viruses 2020, 12, x FOR PEER REVIEW 2 of 3 Supplementary Figure S1. FBS-cultured Huh7.5 choline transporter transcript expression Supplementary Figure S1. FBS-cultured Huh7.5 choline transporter transcript expression. RNA was isolated from FBS-cultured Huh7.5 cells to measure choline transporter (SLC44A1-5; choline transporter-like 1-5, and SLC22A1 and 2; organic cation transporter 1 and 2) transcript expression. All groups are shown relative to the expression of SLC44A1 and normalized to the average of β ACTIN and HSP90. Supplementary Figure S2. HS-cultured Huh7.5 choline uptake kinetics Supplementary Figure S2. HS-cultured Huh7.5 choline uptake kinetics. Choline uptake saturation kinetics from HS-cultured Huh7.5 cells, fit to a Michaelis-Menten curve. Data are mean±SEM and are representative of 3 independent experiments. Viruses 2020, 12, x FOR PEER REVIEW 3 of 3 Supplementary Figure S3. Inhibition of choline transport in FBS-cultured infected cells Supplementary Figure S3. Inhibition of choline transport in FBS-cultured infected cells. FBS-cultured Huh7.5 cells were infected at an MOI of 1 for 24 h in the presence or absence of 20 or 200 μM hemicholinium-3 (HC-3) to inhibit choline uptake. -

Control of Choline Oxidation in Rat Kidney Mitochondria
ÔØ ÅÒÙ×Ö ÔØ Control of Choline Oxidation in Rat Kidney Mitochondria Niaobh O’Donoghue, Trevor Sweeney, Robin Donagh, Kieran Clarke, Richard K. Porter PII: S0005-2728(09)00144-3 DOI: doi:10.1016/j.bbabio.2009.04.014 Reference: BBABIO 46295 To appear in: BBA - Bioenergetics Received date: 11 March 2009 Revised date: 27 April 2009 Accepted date: 29 April 2009 Please cite this article as: Niaobh O’Donoghue, Trevor Sweeney, Robin Donagh, Kieran Clarke, Richard K. Porter, Control of Choline Oxidation in Rat Kidney Mitochondria, BBA - Bioenergetics (2009), doi:10.1016/j.bbabio.2009.04.014 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. ACCEPTED MANUSCRIPT Control of Choline Oxidation in Rat Kidney Mitochondria Niaobh O’Donoghue, Trevor Sweeney, Robin Donagh, Kieran Clarke and Richard K. Porter * School of Biochemistry and Immunology, Trinity College Dublin, Dublin 2 Ireland. *Correspondence to: Richard K. Porter, School of Biochemistry and Immunology, Trinity College Dublin, Dublin 2 Ireland. Tel. +353-1-8961617; Fax +353-1-6772400; Email: [email protected] Key words: choline, betaine, mitochondria, osmolyte, kidney, transporter ACCEPTED MANUSCRIPT Abbreviations: EGTA, ethylenebis(oxethylenenitrilo)tetraacetic acid; FCCP, carbonyl cyanide p-trifluoromethoxyphenylhyrazone; HEPES, 4-(2- hydroxyethyl)-1-peiperazine-ethanesulfonic acid; MOPS, (3-[N}- morphilino)propane sulphonic acid; TPMP, methyltriphenylphosphonium 1 ACCEPTED MANUSCRIPT ABSTRACT Choline is a quaternary amino cationic organic alcohol that is oxidized to betaine in liver and kidney mitochondria. -

Sodium-Coupled Glucose Transport, the SLC5 Family, and Therapeutically Relevant Inhibitors: from Molecular Discovery to Clinical Application
Pflügers Archiv - European Journal of Physiology (2020) 472:1177–1206 https://doi.org/10.1007/s00424-020-02433-x INVITED REVIEW Sodium-coupled glucose transport, the SLC5 family, and therapeutically relevant inhibitors: from molecular discovery to clinical application Gergely Gyimesi1 & Jonai Pujol-Giménez1 & Yoshikatsu Kanai2 & Matthias A. Hediger1 Received: 4 March 2020 /Revised: 24 June 2020 /Accepted: 2 July 2020 / Published online: 7 August 2020 # The Author(s) 2020 Abstract Sodium glucose transporters (SGLTs) belong to the mammalian solute carrier family SLC5. This family includes 12 different members in human that mediate the transport of sugars, vitamins, amino acids, or smaller organic ions such as choline. The SLC5 family belongs to the sodium symporter family (SSS), which encompasses transporters from all kingdoms of life. It furthermore shares similarity to the structural fold of the APC (amino acid-polyamine-organocation) transporter family. Three decades after the first molecular identification of the intestinal Na+-glucose cotransporter SGLT1 by expression cloning, many new discoveries have evolved, from mechanistic analysis to molecular genetics, structural biology, drug discovery, and clinical applications. All of these advances have greatly influenced physiology and medicine. While SGLT1 is essential for fast absorption of glucose and galactose in the intestine, the expression of SGLT2 is largely confined to the early part of the kidney proximal tubules, where it reabsorbs the bulk part of filtered glucose. SGLT2 has been successfully exploited by the pharmaceutical industry to develop effective new drugs for the treatment of diabetic patients. These SGLT2 inhibitors, termed gliflozins, also exhibit favorable nephroprotective effects and likely also cardioprotective effects. -

Choline Transport and Metabolism in Genetically Deficient and Chronic Disease States
Choline Transport and Metabolism in Genetically Deficient and Chronic Disease States by Laila Cigana Schenkel A Thesis presented to The University of Guelph In partial fulfilment of requirements for the degree of Doctor of Philosophy in Human Health and Nutritional Sciences Guelph, Ontario, Canada © Laila Cigana Schenkel, August, 2014 ABSTRACT CHOLINE TRANSPORT AND METABOLISM IN GENETICALLY DEFICIENT AND CHRONIC DISEASE STATES Laila Cigana Schenkel Advisor: University of Guelph, 2014 Marica Bakovic Choline is required for the biosynthesis of phosphatidylcholine (PC) by the CDP-choline Kennedy pathway and of betaine. Choline also plays a role in lipid metabolism and hepatic and muscle function. The availability of intracellular choline is regulated by the choline transporter CTL1/SLC44A1 at the plasma membrane. This thesis aims to elucidate the effect of metabolic altered states, such as lipid overload, choline deficiency and CDP:phopshoethanolamine cytidylyltransferase (Pcyt2) genetic modified models, on the choline transport and metabolism. First, we investigated the effect of high fatty acid supply in C2C12 muscle cells. Palmitic acid (PAM) reduced total and plasma membrane CTL1/SLC44A1 protein by activating lysosomal degradation, and limited the choline uptake while increasing diacylglycerol (DAG) and triacylglycerol (TAG) synthesis. Oleic acid (OLA) maintained total and plasma membrane CTL1/SLC44A1, increasing PC and TAG synthesis more than PAM, which offers a protection mechanism from the excess of intracellular DAG and autophagy. We next characterized the choline metabolic defect in fibroblast cells isolated from a Postural Tachycardia Syndrome (POTS) patient, who had low plasma choline. In the POTS fibroblasts, the CTL1/SLC44A1 expression and choline uptake were decreased. PC synthesis and the phospholipid pool were reduced these cells compared to control. -

Lethal Impairment of Cholinergic Neurotransmission in Hemicholinium-3-Sensitive Choline Transporter Knockout Mice
Lethal impairment of cholinergic neurotransmission in hemicholinium-3-sensitive choline transporter knockout mice Shawn M. Ferguson*, Mihaela Bazalakova*, Valentina Savchenko†, Juan Carlos Tapia‡, Jane Wright†, and Randy D. Blakely*†§¶ *Neuroscience Graduate Program and †Department of Pharmacology, Vanderbilt University, Nashville, TN 37232; ‡Department of Anatomy and Neurobiology, Washington University, St. Louis, MO 63110; and §Center for Molecular Neuroscience, Vanderbilt University School of Medicine, Nashville, TN 37232-8548 Edited by Fred H. Gage, The Salk Institute for Biological Studies, San Diego, CA, and approved April 27, 2004 (received for review March 9, 2004) Presynaptic acetylcholine (ACh) synthesis and release is thought to aptic terminals (15, 16, 18). Indeed, the dynamic regulation of be sustained by a hemicholinium-3-sensitive choline transporter HACU in response to changes in cholinergic neuronal activity CHT). We disrupted the murine CHT gene and examined CHT؊͞؊ appears to match presynaptic ACh synthesis to the rate of ACh) .(and ؉͞؊ animals for evidence of impaired cholinergic neurotrans- release (19 mission. Although morphologically normal at birth, CHT؊͞؊ mice Despite several decades of research on presynaptic cholinergic become immobile, breathe irregularly, appear cyanotic, and die mechanisms, the identity of the choline transporter (CHT) has within an hour. Hemicholinium-3-sensitive choline uptake and only recently become clear (20). Okuda and coworkers (21), as ؊͞؊ subsequent ACh synthesis are specifically lost in CHT mouse well as our own group (22, 23), have shown that the transfection brains. Moreover, we observe a time-dependent loss of spontane- of cloned CHT cDNAs elicits Naϩ͞ClϪ-dependent HC-3- ؊͞؊ ous and evoked responses at CHT neuromuscular junctions. -

Vesicular and Plasma Membrane Transporters for Neurotransmitters
Downloaded from http://cshperspectives.cshlp.org/ on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press Vesicular and Plasma Membrane Transporters for Neurotransmitters Randy D. Blakely1 and Robert H. Edwards2 1Department of Pharmacology and Psychiatry, Vanderbilt University School of Medicine, Nashville, Tennessee 37232-8548 2Departments of Neurology and Physiology, UCSF School of Medicine, San Francisco, California 94143 Correspondence: [email protected] The regulated exocytosis that mediates chemical signaling at synapses requires mechanisms to coordinate the immediate response to stimulation with the recycling needed to sustain release. Two general classes of transporter contribute to release, one located on synaptic ves- icles that loads them with transmitter, and a second at the plasma membrane that both termi- nates signaling and serves to recycle transmitter for subsequent rounds of release. Originally identified as the target of psychoactive drugs, these transport systems have important roles in transmitter release, but we are only beginning to understand their contribution to synaptic transmission, plasticity, behavior, and disease. Recent work has started to provide a structural basis for their activity, to characterize their trafficking and potential for regulation. The results indicate that far from the passive target of psychoactive drugs, neurotransmitter transporters undergo regulation that contributes to synaptic plasticity. he speed and potency of synaptic transmis- brane, more active reuptake should help to re- Tsion depend on the immediate availability plenish the pool of releasable transmitter, but of synaptic vesicles filled with high concentra- may also reduce the extent and duration of sig- tions of neurotransmitter. In this article, we fo- naling to the postsynaptic cell. -

Transporters
University of Dundee The Concise Guide to PHARMACOLOGY 2015/16 Alexander, Stephen P. H.; Kelly, Eamonn; Marrion, Neil; Peters, John A.; Benson, Helen E.; Faccenda, Elena Published in: British Journal of Pharmacology DOI: 10.1111/bph.13355 Publication date: 2015 Licence: CC BY Document Version Publisher's PDF, also known as Version of record Link to publication in Discovery Research Portal Citation for published version (APA): Alexander, S. P. H., Kelly, E., Marrion, N., Peters, J. A., Benson, H. E., Faccenda, E., Pawson, A. J., Sharman, J. L., Southan, C., Davies, J. A., & CGTP Collaborators (2015). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. British Journal of Pharmacology, 172(24), 6110-6202. https://doi.org/10.1111/bph.13355 General rights Copyright and moral rights for the publications made accessible in Discovery Research Portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from Discovery Research Portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain. • You may freely distribute the URL identifying the publication in the public portal. Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Download date: 06. Oct. 2021 S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2015/16: Transporters.