Optimizing and Applying Graphene As a Saturable Absorber For
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Demonstration of Inp-On-Si Self-Pulsating DFB Laser Diodes for Optical Microwave Generation Volume 9, Number 4, August 2017
Open Access Demonstration of InP-on-Si Self-Pulsating DFB Laser Diodes for Optical Microwave Generation Volume 9, Number 4, August 2017 Keqi Ma M. Shahin, Student Member, IEEE A. Abbasi, Member, IEEE G. Roelkens, Member, IEEE G. Morthier, Senior Member, IEEE DOI: 10.1109/JPHOT.2017.2724840 1943-0655 © 2017 IEEE IEEE Photonics Journal Demonstration of InP-on-Si Self-Pulsating DFB Laser Demonstration of InP-on-Si Self-Pulsating DFB Laser Diodes for Optical Microwave Generation Keqi Ma,1 M. Shahin,2,3 Student Member, IEEE, A. Abbasi,2,3 Member, IEEE, G. Roelkens,2,3 Member, IEEE, and G. Morthier,2,3 Senior Member, IEEE 1Centre for Optical and Electromagnetic Research, State Key Laboratory for Modern Optical Instrumentation, Zhejiang University, Hangzhou 310027, China 2Photonics Research Group, Department of Information Technology (INTEC), Ghent University–IMEC, Gent B-9000, Belgium 3Center for Nano-and Biophotonics, Ghent University, Gent B-9000, Belgium DOI:10.1109/JPHOT.2017.2724840 1943-0655 C 2017 IEEE. Translations and content mining are permitted for academic research only. Personal use is also permitted, but republication/redistribution requires IEEE permission. See http://www.ieee.org/publications_standards/publications/rights/index.html for more information. Manuscript received June 9, 2017; revised July 5, 2017; accepted July 5, 2017. Date of publication July 11, 2017; date of current version July 26, 2017. This work was supported by the Methusalem program of the Flemish Government. (Keqi Ma and M. Shahin contributed equally to this work.) Corresponding author: M. Shahin (e-mail: [email protected]). Abstract: We demonstrate self-pulsating heterogeneously integrated III-V-on-SOI two- section distributed feedback laser diodes, with a wide range of self-pulsation frequencies between (but not limited to) 12 and 40 GHz. -

Short Pulse Generation Using Multisegment Mode-Locked
2186 IEEE JOURNAL OF QUANTUM ELECTRONICS, VOL. 28, NO. 10, OCTOBER 1992 Short Pulse Generation Using Multisegment - . * T Mode-Locked Semconductor Lasers Dennis J. Derickson, Member, IEEE, Roger J. Helkey, Member, IEEE, Alan Mar, Judy R. Karin, Student Member, IEEE, John G. Wasserbauer, Student Member, IEEE, and John E. Bowers, Senior Member, IEEE Invited Paper Abstract-Mode-locked semiconductor lasers which incorpo- SectionC SedionB SeetionA rate multiple contacting segments are found to give improved performance over single-segment designs. The functions of gain, saturable absorption, gain modulation, repetition rate tuning, wavelength tuning, and electrical pulse generation can be integrated on a single semiconductor chip. The optimization of the performance of multisegment mode-locked lasers in terms va 7F of material parameters, waveguiding parameters, electrical parasitics, and segment length is discussed experimentally and theoretically. ExternalI Cavity I. INTRODUCTION EMICONDUCTOR lasers are important sources of Sshort optical and electrical pulses. Semiconductor la- sers are small in size, use electrical pumping, are easy to operate, and consequently short optical pulses can now be (b) used in applications that were previously infeasible or un- SedionC SedionA economical. These pulses are used in high-speed optical fiber communication systems using time-division multi- Gain-Switch plexing for transmitters and for demultiplexing at the re- Q-SllitCh ceiver [l], [2]. Short optical pulses are also being used for optoelectronic measurement applications such as elec- trooptic sampling [3], analog to digital (A-D) conversion [4], and impulse response testing of optical components. (C) This paper concentrates on advances in the short pulse Fig. 1. Multisegment structures for generating short optical and electrical generation field that are a result of using multiple-segment pulses. -
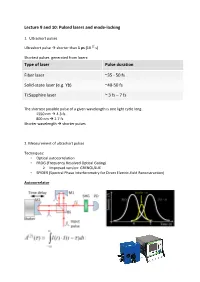
Lecture 9 and 10: Pulsed Lasers and Mode-Locking
Lecture 9 and 10: Pulsed lasers and mode-locking 1. Ultrashort pulses Ultrashort pulse shorter than 1 ps (10-12 s) Shortest pulses generated from lasers: Type of laser Pulse duration Fiber laser ~35 - 50 fs Solid-state laser (e.g. Yb) ~40-50 fs Ti:Sapphire laser ~ 3 fs – 7 fs The shortest possible pulse of a given wavelength is one light cycle long: 1550 nm 4.3 fs 800 nm 2.7 fs Shorter wavelength shorter pulses 2. Measurement of ultrashort pulses Techniques: • Optical autocorrelation • FROG (Frequency Resolved Optical Gating) Improved version: GRENOULLIE • SPIDER (Spectral Phase Interferometry for Direct Electric-field Reconstruction) Autocorrelator Interferometric autocorrelation trace: FROG Source: Rick Trebino, Frequency Resolved Optical Gating SPIDER 3. Mode-locked lasers a) Difference between a CW and mode-locked laser: A mode-locked laser: Emits a train of equally-spaced, ultrashort pulses (femtosecond) - down to almost single- cycle Output spectrum is broad (tens-hundreds of nanometers)/ Broader spectrum shorter pulses In the frequency domain, the output consists of thousands/millions of narrow lines (comb- like structure) b) Relation between the number of synchronized modes and the pulse duration: More synchronized modes shorter pulse. c) Passive mode-locking and saturable absorber In order to achieve passive mode-locking we need a device called saturable absorber. The saturable absorber: - Blocks any CW radiation in the cavity - It forces the laser to synchronize the modes and generate pulses - Only high-intensity pulse -

Mid-Infrared Ultra-Short Mode-Locked Fiber Laser Utilizing Topological
Mid-infrared ultra-short mode-locked fiber laser utilizing topological insulator BiR2RTeR3R nano-sheets as the saturable absorber 1 1,2,3 1 1 1,2 1,4 Ke YinP ,P Tian JiangP ,P Hao YuP ,P Xin ZhengP ,P Xiangai ChengP ,P Jing HouP 1 P CollegeP of Optoelectronic Science and Engineering, National University of Defense Technology, Changsha 410073, P.R.China; 2 P StateP key laboratory of High Performance Computing, National University of Defense Technology, Changsha 410073, P.R.China; 3 P P [email protected] U 4 PU hUP [email protected] Abstract: The newly-emergent two-dimensional topological insulators (TIs) have shown their unique electronic and optical properties, such as good thermal management, high nonlinear refraction index and ultrafast relaxation time. Their narrow energy band gaps predict their optical absorption ability further into the mid-infrared region and their possibility to be very broadband light modulators ranging from the visible to the mid-infrared region. In this paper, a mid-infrared mode-locked fluoride fiber laser with TI Bi2Te3 nano-sheets as the saturable absorber is presented. Continuous wave lasing, Q-switched and continuous-wave mode-locking (CW-ML) operations of the laser are observed sequentially by increasing the pump power. The observed CW-ML pulse train has a pulse repetition rate of 10.4 MHz, a pulse width of ~6 ps, and a center wavelength of 2830 nm. The maximum achievable pulse energy is 8.6 nJ with average power up to 90 mW. This work forcefully demonstrates the promising applications of two-dimensional TIs for ultra-short laser operation and nonlinear optics in the mid-infrared region. -

Numerical Simulation of Passively Q-Switched Solid State Lasers
Chapter 14 Numerical Simulation of Passively Q-Switched Solid State Lasers I. Lăncrănjan, R. Savastru, D. Savastru and S. Micloş Additional information is available at the end of the chapter http://dx.doi.org/10.5772/47812 1. Introduction Short laser light pulses have a large number of applications in many civilian and military applications [1-10]. To a large percentage these short laser pulses are generated by solid state lasers using various active media types (crystals, glasses or ceramic) operated in Q- switching and/or mode-locking techniques [1-10]. Among the short light pulses laser generators, those operated in Q-switching regime and emitting pulses of nanosecond FWHM duration occupy a large part of civilian (material processing - for example: nano- materials formation by using ablation technique [3,7]) and military (projectile guidance over long distances and range finding) applications. Basically, Q-switching operation relies on a fast switching of laser resonator quality factor Q from a low value (corresponding to large optical losses) to a high one (representing low radiation losses). Depending on the proposed application, two main Q-switching techniques are used: active, based on electrical (in some cases operation with high voltages up to about 1 kV being necessary) or mechanical (spinning speeds up to about 1 kHz being used) actions on an optical component at least, coming from the outside of the laser resonator, and passive relying entirely on internal to the laser resonator induced variation of one optical component transmittance. 2. Theory Figure 1 displays a general schematic of electronic energy levels of laser active centers embedded in an active medium and of saturable absorption centers embedded into solid state passive optical Q-switch cell laser oscillator operated in passive optical Q-switching regime [11-18,26]. -

Advances in All-Solid-State Passively Q-Switched Lasers Based on Cr4+:YAG Saturable Absorber
hv photonics Review Advances in All-Solid-State Passively Q-Switched Lasers Based on Cr4+:YAG Saturable Absorber Jingling Tang 1,2, Zhenxu Bai 1,2,3,*, Duo Zhang 1,2, Yaoyao Qi 1,2, Jie Ding 1,2, Yulei Wang 1,2 and Zhiwei Lu 1,2 1 Center for Advanced Laser Technology, Hebei University of Technology, Tianjin 300401, China; [email protected] (J.T.); [email protected] (D.Z.); [email protected] (Y.Q.); [email protected] (J.D.); [email protected] (Y.W.); [email protected] (Z.L.) 2 Hebei Key Laboratory of Advanced Laser Technology and Equipment, Tianjin 300401, China 3 MQ Photonics Research Centre, Department of Physics and Astronomy, Macquarie University, Sydney, NSW 2109, Australia * Correspondence: [email protected] Abstract: All-solid-state passively Q-switched lasers have advantages that include simple structure, high peak power, and short sub-nanosecond pulse width. Potentially, these lasers can be applied in multiple settings, such as in miniature light sources, laser medical treatment, remote sensing, and precision processing. Cr4+:YAG crystal is an ideal Q-switch material for all-solid-state passively Q-switched lasers owing to its high thermal conductivity, low saturation light intensity, and high damage threshold. This study summarizes the research progress on all-solid-state passively Q- switched lasers that use Cr4+:YAG crystal as a saturable absorber and discusses further prospects for the development and application of such lasers. Keywords: laser; Cr4+:YAG; all-solid-state; passively Q-switch Citation: Tang, J.; Bai, Z.; Zhang, D.; Qi, Y.; Ding, J.; Wang, Y.; Lu, Z. -

Mechanisms of Spatiotemporal Mode-Locking
Mechanisms of Spatiotemporal Mode-Locking Logan G. Wright1, Pavel Sidorenko1, Hamed Pourbeyram1, Zachary M. Ziegler1, Andrei Isichenko1, Boris A. Malomed2,3, Curtis R. Menyuk4, Demetrios N. Christodoulides5, and Frank W. Wise1 1. School of Applied and Engineering Physics, Cornell University, Ithaca, NY 14853, USA 2. Department of Physical Electronics, School of Electrical Engineering, Faculty of Engineering, and the Center for Light-Matter Interaction, Tel Aviv University, 69978 Tel Aviv, Israel 3. ITMO University, St. Petersburg 197101, Russia 4. Department of Computer Science and Electrical Engineering, University of Maryland Baltimore County, Baltimore, Maryland 21250, USA 5. CREOL/College of Optics and Photonics, University of Central Florida, Orlando, Florida 32816, USA Abstract Mode-locking is a process in which different modes of an optical resonator establish, through nonlinear interactions, stable synchronization. This self-organization underlies light sources that enable many modern scientific applications, such as ultrafast and high-field optics and frequency combs. Despite this, mode-locking has almost exclusively referred to self-organization of light in a single dimension - time. Here we present a theoretical approach, attractor dissection, for understanding three-dimensional (3D) spatiotemporal mode-locking (STML). The key idea is to find, for each distinct type of 3D pulse, a specific, minimal reduced model, and thus to identify the important intracavity effects responsible for its formation and stability. An intuition for the results follows from the “minimum loss principle,” the idea that a laser strives to find the configuration of intracavity light that minimizes loss (maximizes gain extraction). Through this approach, we identify and explain several distinct forms of STML. -

Atomic Layer Graphene As Saturable Absorber for Ultrafast Pulsed Lasers **
Atomic layer graphene as saturable absorber for ultrafast pulsed lasers ** By Qiaoliang Bao, Han Zhang, Yu Wang, Zhenhua Ni, Yongli Yan, Ze Xiang Shen, Kian Ping Loh*,and Ding Yuan Tang* [*] Prof. K. P. Loh. Corresponding-Author, Dr. Q. Bao, Dr. Y. Wang, Dr. Y. Yan Department of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543 (Singapore) E-mail: [email protected] Prof. D. Y. Tang, H. Zhang School of Electrical and Electronic Engineering, Nanyang Technological University, Singapore 639798 (Singapore) E-mail: [email protected] Prof. Z. X. Shen, Dr. Z. Ni School of Physical and Mathematical Sciences, Nanyang Technological University Singapore 639798 (Singapore) [**] This project is funded by NRF-CRP award “Graphene and Related Materials and Devices”. Supporting Information is available online from Wiley InterScience or from the author. ٛٛ1 Keywords: Graphene, Nonlinear Optics, Photonics Abstract The optical conductance of monolayer graphene is defined solely by the fine structure constant, α = e2/ћc (where e is the electron charge, ћ is Dirac’s constant and c is the speed of light). The absorbance has been predicted to be independent of frequency. In principle, the interband optical absorption in zero-gap graphene could be saturated readily under strong excitation due to Pauli blocking. Here, we demonstrate the use of atomic layer graphene as saturable absorber in a mode-locked fiber laser for the generation of ultrashort soliton pulses (756 fs) at the telecommunication band. The modulation depth can be tuned in a wide range from 66.5% to 6.2% by varying the thickness of graphene. -

Passively Q-Switched Erbium-Doped Fiber Laser Using Fe3o4-Nanoparticle Saturable Absorber
Passively Q-switched Erbium-doped fiber laser using Fe3O4-nanoparticle saturable absorber Xuekun Bai,1,a Chengbo Mou,2,a Luxi Xu,1 Sujuan Huang,1 Tingyun Wang,1 Shengli Pu,3 Xianglong Zeng1,* 1The Key Lab of Specialty Fiber Optics and Optical Access Network, Shanghai University, 200072 Shanghai, China 2 Aston Institute of Photonic Technologies (AIPT), Aston University, Aston Triangle, Birmingham B4 7ET, United Kingdom 3 College of science, University of Shanghai for Science and Technology, 200093 Shanghai, China aThese authors contribute equally to the work *Corresponding author: [email protected] Received Month X, XXXX; revised Month X, XXXX; accepted Month X, XXXX; posted Month X, XXXX (Doc. ID XXXXX); published Month X, XXXX We experimentally demonstrate a passively Q-switched erbium-doped fiber laser (EDFL) operation by using a saturable absorber based on Fe3O4 nanoparticles (FONP) in magnetic fluid (MF). As a kind of transition metal oxide, the FONP has a large nonlinear optical response with a fast response time for saturable absorber. By depositing MF at the end of optical fiber ferrule, we fabricated a FONP-based saturable absorber, which enables a strong light-matter interaction owing to the confined transmitted optical field within the single mode fiber. Because of large third-order optical nonlinearities of FONP-based saturable absorber, large modulation depth of 8.2% and non saturable absorption of 56.6% are demonstrated. As a result, stable passively Q-switched EDFL pulses with maximum output pulse energy of 23.76 nJ, repetition rate of 33.3 kHz, and pulse width of 3.2 μs are achieved when the input pump power is 110 mW at the wavelength of 980 nm. -
Passively Q-Switched Ytterbium-Doped Fiber Laser
www.nature.com/scientificreports OPEN Passively Q-switched Ytterbium- doped fber laser based on broadband multilayer Platinum Received: 13 March 2019 Accepted: 3 July 2019 Ditelluride (PtTe2) saturable Published: xx xx xxxx absorber Ping Kwong Cheng1,2, Chun Yin Tang1,2, Xin Yu Wang1,2, Sainan Ma1,2, Hui Long1,2 & Yuen Hong Tsang 1,2 Two-dimensional (2D) layered Platinum Ditelluride (PtTe2), a novel candidate of group 10 transition- metal dichalcogenides (TMDs), which provides enormous potential for pulsed laser applications due to its highly stable and strong nonlinear optical absorption (NOA) properties. PtTe2 saturable absorber (SA) is successfully fabricated with frstly demonstrated the passively Q-switched laser operation within a Yb-doped fber laser cavity at 1066 nm. Few layered PtTe2 is produced by uncomplicated and cost- efcient ultrasonic liquid exfoliation and follow by incorporating into polyvinyl alcohol (PVA) polymer to form a PtTe2-PVA composite thin flm saturable absorber. The highest achieved single pulse energy is 74.0 nJ corresponding to pulse duration, repetition rate and average output power of 5.2 μs, 33.5 kHz and 2.48 mW, respectively. This work has further exploited the immeasurable utilization potential of the air stable and broadband group 10 TMDs for ultrafast photonic applications. Q-switching, a useful and very important technique, has been widely studied and applied in pulsed laser devel- opment over the past decades1–4. Contributed by the remarkable high pulse peak power, Q-switched laser can be employed in vast applications, for instance, industrial laser engraving, nonlinear optics, skin treatment, and eyes surgery5–7. Q-switched laser pulses can be produced by using Acousto-optical or Electro-optical modulators (AOM/EOM)8 to actively modify the Quality factor within the cavity. -
Spectral Dynamics on Saturable Absorber in Mode-Locking with Time Stretch Spectroscopy
UC Irvine UC Irvine Previously Published Works Title Spectral dynamics on saturable absorber in mode-locking with time stretch spectroscopy. Permalink https://escholarship.org/uc/item/3s63r63k Journal Scientific reports, 10(1) ISSN 2045-2322 Authors Suzuki, Masayuki Boyraz, Ozdal Asghari, Hossein et al. Publication Date 2020-09-02 DOI 10.1038/s41598-020-71342-x Peer reviewed eScholarship.org Powered by the California Digital Library University of California www.nature.com/scientificreports OPEN Spectral dynamics on saturable absorber in mode‑locking with time stretch spectroscopy Masayuki Suzuki1*, Ozdal Boyraz2, Hossein Asghari3 & Bahram Jalali4 A mode‑locked laser that can produce a broadband spectrum and ultrashort pulse has been applied for many applications in an extensive range of scientifc felds. To obtain stable mode-locking during a long time alignment-free, a semiconductor saturable absorber is one of the most suitable devices. Dynamics from noise to a stable mode‑locking state in the spectral‑domain are known as complex and a non-repetitive phenomenon with the time scale from nanoseconds to milliseconds. Thus, a conventional spectrometer, which is composed of a grating and line sensor, cannot capture the spectral behavior from noise to stable mode-locking. As a powerful spectral measurement technique, a time‑stretch dispersive Fourier transformation (TS‑DFT) has been recently used to enable a successive single-shot spectral measurement over a couple of milliseconds time span. Here, we experimentally demonstrate real‑time spectral evolution of femtosecond pulse build‑up in a homemade passive mode-locked Yb fber laser with a semiconductor saturable absorber mirror using TS-DFT. -

High-Quality, Inn-Based, Saturable Absorbers for Ultrafast Laser Development
applied sciences Review High-Quality, InN-Based, Saturable Absorbers for Ultrafast Laser Development Laura Monroy 1,* , Marco Jiménez-Rodríguez 1, Eva Monroy 2 , Miguel González-Herráez 1 and Fernando B. Naranjo 1 1 Grupo de Ingeniería Fotónica, Departamento de Electrónica (EPS) Universidad de Alcalá, Campus Universitario, Alcalá de Henares, 28871 Madrid, Spain; [email protected] (M.J.-R.); [email protected] (M.G.-H.); [email protected] (F.B.N.) 2 CEA-IRIG-DEPHY-PHELIQS, Univ. Grenoble-Alpes, 17 av. des Martyrs, Grenoble 38000, France; [email protected] * Correspondence: [email protected]; Tel.: +64-91-885-58-97 Received: 24 September 2020; Accepted: 3 November 2020; Published: 4 November 2020 Abstract: New fabrication methods are strongly demanded for the development of thin-film saturable absorbers with improved optical properties (absorption band, modulation depth, nonlinear optical response). In this sense, we investigate the performance of indium nitride (InN) epitaxial layers 18 3 with low residual carrier concentration (<10 cm− ), which results in improved performance at telecom wavelengths (1560 nm). These materials have demonstrated a huge modulation depth of 23% and a saturation fluence of 830 µJ/cm2, and a large saturable absorption around 3 104 cm/GW − × has been observed, attaining an enhanced, nonlinear change in transmittance. We have studied the use of such InN layers as semiconductor saturable absorber mirrors (SESAMs) for an erbium (Er)-doped fiber laser to perform mode-locking generation at 1560 nm. We demonstrate highly stable, ultrashort (134 fs) pulses with an energy of up to 5.6 nJ. Keywords: saturable absorbers; nonlinear effects; material defects 1.