BMC Genomics Biomed Central
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ATAP00021-Recombinant Human ALDH1A1 Protein
ATAGENIX LABORATORIES Catalog Number:ATAP00021 Recombinant Human ALDH1A1 protein Product Details Summary English name Recombinant Human ALDH1A1 protein Purity >90% as determined by SDS-PAGE Endotoxin level Please contact with the lab for this information. Construction A DNA sequence encoding the human ALDH1A1 (Met1-Ser501) was fused with His tag Accession # P00352 Host E.coli Species Homo sapiens (Human) Predicted Molecular Mass 52.58 kDa Formulation Supplied as solution form in PBS pH 7.5 or lyophilized from PBS pH 7.5. Shipping In general, proteins are provided as lyophilized powder/frozen liquid. They are shipped out with dry ice/blue ice unless customers require otherwise. Stability &Storage Use a manual defrost freezer and avoid repeated freeze thaw cycles. Store at 2 to 8 °C for one week . Store at -20 to -80 °C for twelve months from the date of receipt. Reconstitution Reconstitute in sterile water for a stock solution.A copy of datasheet will be provided with the products, please refer to it for details. Background Background Aldehyde dehydrogenase 1 family, member A1 (ALDH1A1), also known as Aldehyde dehydrogenase 1 (ALDH1), or Retinaldehyde Dehydrogenase 1 (RALDH1), is an enzyme that is expressed at high levels in stem cells and that has been suggested to regulate stem cell function. The retinaldehyde dehydrogenase (RALDH) subfamily of ALDHs, composed of ALDH1A1, ALDH1A2, ALDH1A3, and ALDH8A1, regulate development by catalyzing retinoic acid biosynthesis. The ALDH1A1 protein belongs to the aldehyde dehydrogenases family of proteins. Aldehyde dehydrogenase is the second enzyme of the major oxidative pathway of alcohol metabolism. ALDH1A1 also belongs to the group of corneal crystallins that Web:www.atagenix.com E-mail: [email protected] Tel: 027-87433958 ATAGENIX LABORATORIES Catalog Number:ATAP00021 Recombinant Human ALDH1A1 protein help maintain the transparency of the cornea. -

Aldehyde Dehydrogenase, Liver Disease and Cancer Wenjun Wang1, Chunguang Wang2, Hongxin Xu1, Yanhang Gao1
Int. J. Biol. Sci. 2020, Vol. 16 921 Ivyspring International Publisher International Journal of Biological Sciences 2020; 16(6): 921-934. doi: 10.7150/ijbs.42300 Review Aldehyde Dehydrogenase, Liver Disease and Cancer Wenjun Wang1, Chunguang Wang2, Hongxin Xu1, Yanhang Gao1 1. Department of Hepatology, The First Hospital of Jilin University, Jilin University, Changchun, Jilin, 130021, China. 2. Department of Thoracic & Cardiovascular Surgery, Second Clinical College, Jilin University, Changchun, 130041, China. Corresponding author: Yanhang Gao, MD., PhD., Department of Hepatology, The First Hospital of Jilin University, Jilin University, Changchun, Jilin, 130021, China. Email: [email protected]. Tel: +86 15804303019; +86 431 81875121; +86 431 81875106; Fax number: 0431-81875106. © The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See http://ivyspring.com/terms for full terms and conditions. Received: 2019.11.20; Accepted: 2020.01.03; Published: 2020.01.22 Abstract Acetaldehyde dehydrogenase 2 (ALDH2) is the key enzyme responsible for metabolism of the alcohol metabolite acetaldehyde in the liver. In addition to conversion of the acetaldehyde molecule, ALDH is also involved in other cellular functions. Recently, many studies have investigated the involvement of ALDH expression in viral hepatitis, alcoholic liver disease (ALD), non-alcoholic fatty liver disease (NAFLD), liver fibrosis, and liver cancer. Notably, ALDH2 expression has been linked with liver cancer risk, as well as pathogenesis and prognosis, and has emerged as a promising therapeutic target. Of note, approximately 8% of the world’s population, and approximately 30-40% of the population in East Asia carry an inactive ALDH2 gene. -

The Cancer Stem Cell Marker Aldehyde Dehydrogenase Is Required to Maintain a Drug-Tolerant Tumor Cell Subpopulation
Published OnlineFirst May 8, 2014; DOI: 10.1158/0008-5472.CAN-13-3456 Cancer Therapeutics, Targets, and Chemical Biology Research The Cancer Stem Cell Marker Aldehyde Dehydrogenase Is Required to Maintain a Drug-Tolerant Tumor Cell Subpopulation Debasish Raha1, Timothy R. Wilson1, Jing Peng2, David Peterson2, Peng Yue3, Marie Evangelista1, Catherine Wilson1, Mark Merchant2, and Jeff Settleman1 Abstract Selective kinase inhibitors have emerged as an important class of cancer therapeutics, and several such drugs are now routinely used to treat advanced-stage disease. However, their clinical benefit is typically short-lived because of the relatively rapid acquisition of drug resistance following treatment response. Accumulating preclinical and clinical data point to a role for a heterogeneous response to treatment within a subpopulation of tumor cells that are intrinsically drug-resistant, such as cancer stem cells. We have previously described an epigenetically determined reversibly drug-tolerant subpopulation of cancer cells that share some properties with cancer stem cells. Here, we define a requirement for the previously established cancer stem cell marker ALDH (aldehyde dehydrogenase) in the maintenance of this drug-tolerant subpopulation. We find that ALDH protects the drug-tolerant subpopulation from the potentially toxic effects of elevated levels of reactive oxygen species (ROS) in these cells, and pharmacologic disruption of ALDH activity leads to accumulation of ROS to toxic levels, consequent DNA damage, and apoptosis specifically within the drug-tolerant subpopulation. Combining ALDH inhibition with other kinase-directed treatments delayed treatment relapse in vitro and in vivo, revealing a novel combination treatment strategy for cancers that might otherwise rapidly relapse following single-agent therapy. -

Cells in Diabetic Mice
ARTICLE Received 12 Jan 2016 | Accepted 18 Jul 2016 | Published 30 Aug 2016 DOI: 10.1038/ncomms12631 OPEN Aldehyde dehydrogenase 1a3 defines a subset of failing pancreatic b cells in diabetic mice Ja Young Kim-Muller1,*, Jason Fan1,2,*, Young Jung R. Kim2, Seung-Ah Lee1, Emi Ishida1, William S. Blaner1 & Domenico Accili1 Insulin-producing b cells become dedifferentiated during diabetes progression. An impaired ability to select substrates for oxidative phosphorylation, or metabolic inflexibility, initiates progression from b-cell dysfunction to b-cell dedifferentiation. The identification of pathways involved in dedifferentiation may provide clues to its reversal. Here we isolate and functionally characterize failing b cells from various experimental models of diabetes and report a striking enrichment in the expression of aldehyde dehydrogenase 1 isoform A3 (ALDH þ )asb cells become dedifferentiated. Flow-sorted ALDH þ islet cells demonstrate impaired glucose- induced insulin secretion, are depleted of Foxo1 and MafA, and include a Neurogenin3- positive subset. RNA sequencing analysis demonstrates that ALDH þ cells are characterized by: (i) impaired oxidative phosphorylation and mitochondrial complex I, IV and V; (ii) activated RICTOR; and (iii) progenitor cell markers. We propose that impaired mitochondrial function marks the progression from metabolic inflexibility to dedifferentiation in the natural history of b-cell failure. 1 Naomi Berrie Diabetes Center and Department of Medicine, Columbia University, New York, New York 10032, USA. 2 Department of Genetics and Integrated Program in Cellular, Molecular and Biomedical Studies, Columbia University, New York, New York 10032, USA. * These authors contributed equally to this work. Correspondence and requests for materials should be addressed to D.A. -

Metabolic Network-Based Stratification of Hepatocellular Carcinoma Reveals Three Distinct Tumor Subtypes
Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumor subtypes Gholamreza Bidkhoria,b,1, Rui Benfeitasa,1, Martina Klevstigc,d, Cheng Zhanga, Jens Nielsene, Mathias Uhlena, Jan Borenc,d, and Adil Mardinoglua,b,e,2 aScience for Life Laboratory, KTH Royal Institute of Technology, SE-17121 Stockholm, Sweden; bCentre for Host-Microbiome Interactions, Dental Institute, King’s College London, SE1 9RT London, United Kingdom; cDepartment of Molecular and Clinical Medicine, University of Gothenburg, SE-41345 Gothenburg, Sweden; dThe Wallenberg Laboratory, Sahlgrenska University Hospital, SE-41345 Gothenburg, Sweden; and eDepartment of Biology and Biological Engineering, Chalmers University of Technology, SE-41296 Gothenburg, Sweden Edited by Sang Yup Lee, Korea Advanced Institute of Science and Technology, Daejeon, Republic of Korea, and approved November 1, 2018 (received for review April 27, 2018) Hepatocellular carcinoma (HCC) is one of the most frequent forms of of markers associated with recurrence and poor prognosis (13–15). liver cancer, and effective treatment methods are limited due to Moreover, genome-scale metabolic models (GEMs), collections tumor heterogeneity. There is a great need for comprehensive of biochemical reactions, and associated enzymes and transporters approaches to stratify HCC patients, gain biological insights into have been successfully used to characterize the metabolism of subtypes, and ultimately identify effective therapeutic targets. We HCC, as well as identify drug targets for HCC patients (11, 16–18). stratified HCC patients and characterized each subtype using tran- For instance, HCC tumors have been stratified based on the uti- scriptomics data, genome-scale metabolic networks and network lization of acetate (11). Analysis of HCC metabolism has also led topology/controllability analysis. -

Renoprotective Effect of Combined Inhibition of Angiotensin-Converting Enzyme and Histone Deacetylase
BASIC RESEARCH www.jasn.org Renoprotective Effect of Combined Inhibition of Angiotensin-Converting Enzyme and Histone Deacetylase † ‡ Yifei Zhong,* Edward Y. Chen, § Ruijie Liu,*¶ Peter Y. Chuang,* Sandeep K. Mallipattu,* ‡ ‡ † | ‡ Christopher M. Tan, § Neil R. Clark, § Yueyi Deng, Paul E. Klotman, Avi Ma’ayan, § and ‡ John Cijiang He* ¶ *Department of Medicine, Mount Sinai School of Medicine, New York, New York; †Department of Nephrology, Longhua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China; ‡Department of Pharmacology and Systems Therapeutics and §Systems Biology Center New York, Mount Sinai School of Medicine, New York, New York; |Baylor College of Medicine, Houston, Texas; and ¶Renal Section, James J. Peters Veterans Affairs Medical Center, New York, New York ABSTRACT The Connectivity Map database contains microarray signatures of gene expression derived from approximately 6000 experiments that examined the effects of approximately 1300 single drugs on several human cancer cell lines. We used these data to prioritize pairs of drugs expected to reverse the changes in gene expression observed in the kidneys of a mouse model of HIV-associated nephropathy (Tg26 mice). We predicted that the combination of an angiotensin-converting enzyme (ACE) inhibitor and a histone deacetylase inhibitor would maximally reverse the disease-associated expression of genes in the kidneys of these mice. Testing the combination of these inhibitors in Tg26 mice revealed an additive renoprotective effect, as suggested by reduction of proteinuria, improvement of renal function, and attenuation of kidney injury. Furthermore, we observed the predicted treatment-associated changes in the expression of selected genes and pathway components. In summary, these data suggest that the combination of an ACE inhibitor and a histone deacetylase inhibitor could have therapeutic potential for various kidney diseases. -

ALDH1A3 Is the Key Isoform That Contributes to Aldehyde Dehydrogenase Activity and Affects in Vitro Proliferation in Cardiac Atrial Appendage Progenitor Cells
ORIGINAL RESEARCH published: 24 July 2018 doi: 10.3389/fcvm.2018.00090 ALDH1A3 Is the Key Isoform That Contributes to Aldehyde Dehydrogenase Activity and Affects in Vitro Proliferation in Cardiac Atrial Appendage Progenitor Cells Stefania Puttini 1, Isabelle Plaisance 1, Lucio Barile 2, Elisabetta Cervio 2, Giuseppina Milano 1,2, Paola Marcato 3, Thierry Pedrazzini 1 and Giuseppe Vassalli 1,2* 1 Cardiovascular Department, CHUV University Hospital, Lausanne, Switzerland, 2 Cardiocentro Ticino Foundation and Swiss Institute for Regenerative Medicine, Lugano, Switzerland, 3 Departments of Pathology, Microbiology and Immunology, Dalhousie University, Halifax, NS, Canada Edited by: Rajesh Katare, hi University of Otago, New Zealand High aldehyde dehydrogenase (ALDH ) activity has been reported in normal and cancer hi Reviewed by: stem cells. We and others have shown previously that human ALDH cardiac atrial Daniele Torella, appendage cells are enriched with stem/progenitor cells. The role of ALDH in these cells Università degli studi Magna Græcia di Catanzaro, Italy is poorly understood but it may come down to the specific ALDH isoform(s) expressed. Parul Dixit, This study aimed to compare ALDHhi and ALDHlo atrial cells and to identify the isoform(s) Bristol Heart Institute, United Kingdom that contribute to ALDH activity, and their functional role. *Correspondence: Giuseppe Vassalli Methods and Results: Cells were isolated from atrial appendage specimens from [email protected] patients with ischemic and/or valvular heart disease undergoing heart surgery. ALDHhi activity assessed with the Aldefluor reagent coincided with primitive surface marker Specialty section: + hi This article was submitted to expression (CD34 ). Depending on their ALDH activity, RT-PCR analysis of ALDH and Cardiovascular Biologics and ALDHlo cells demonstrated a differential pattern of pluripotency genes (Oct 4, Nanog) Regenerative Medicine, hi a section of the journal and genes for more established cardiac lineages (Nkx2.5, Tbx5, Mef2c, GATA4). -
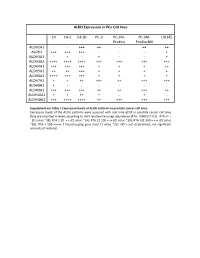
ALDH Expression in Pca Cell Lines C4 C4-2 C4-2B PC-3 PC-3M- Pro4luc PC-3M- Pro4lucbiii DU145 ALDH1A1
ALDH Expression in PCa Cell lines C4 C4-2 C4-2B PC-3 PC-3M- PC-3M- DU145 Pro4luc Pro4lucBIII ALDH1A1 - - +++ ++ - ++ ++ ALDH2 +++ +++ +++ - - - + ALDH3A1 - + - + - - + ALDH3A2 ++++ ++++ ++++ +++ +++ +++ +++ ALDH4A1 +++ +++ +++ + + + ++ ALDH5A1 ++ ++ +++ + + + + ALDH6A1 ++++ +++ +++ + + + + ALDH7A1 + + ++ +++ ++ +++ +++ ALDH8A1 + - - - - - - ALDH9A1 +++ +++ +++ ++ ++ +++ ++ ALDH16A1 + + ++ + - + - ALDH18A1 +++ ++++ ++++ ++ +++ +++ +++ Supplementary Table I. Expression levels of ALDH isoforms in prostate cancer cell lines. Expression levels of the ALDH isoforms were assessed with real time qPCR in prostate cancer cell lines Data are classified in levels according to their relative transcript abundance (RTA: 10000/2^ΔCt). RTA <1 = - (Ct value ~38); RTA 1-20 = + (Ct value ~34); RTA 21-100 = ++ (Ct value ~30); RTA 101-500 = +++ (Ct value ~26); RTA > 500 =++++ ( housekeeping gene level Ct value ~22). ND = not determined, not-significant amounts of material. ALDH Expression in Prostate Cancer PCa primary tumor PCa primary cultures #44 #99 #62#69 123 583 697 233 549 069B 567B ALDH1A1 ++ + ND ND ++++ ND ND ND ND ND ND ALDH2 + ++ ND ND ++++ ND ND ND ND ND ND ALDH3A1 ++++ ++++ +++ ++ ++ +++ ++++ ++++ +++ ++ + ALDH3A2 +++ +++ +++ ++++ + ++++ ++++ ++++ ++++ ++++ ++++ ALDH4A1 ++ +++ ++ ++ +++ ND ND ND ND ND ND ALDH5A1 ++ +++ ++ ++ - ND ND ND ND ND ND ALDH6A1 ++ + ND ND ++++ ND ND ND ND ND ND ALDH7A1 ++++ ++++ +++ +++ ++++ ++++ ++++ ++++ ++++ ++++ ++++ ALDH8A1 + + ND ND + ND ND ND ND ND ND ALDH9A1 ++ ++ ND ND ++++ ND ND ND ND ND ND ALDH16A1 +++ ++ ND ND ++++ ND ND ND ND ND ND ALDH18A1 - - +++ +++ - ++++ ++++ ++++ ++++ +++++++ Supplementary Table II. Expression levels of ALDH isoforms in primary prostate cancer tissue samples and primary cultures. Expression levels of the ALDH isoforms were assessed with real time qPCR in primary prostate cancer tissue samples (left panel) and primary cultures (right panel). Data are classified in levels according to their relative transcript abundance. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

Supplemental Figures 04 12 2017
Jung et al. 1 SUPPLEMENTAL FIGURES 2 3 Supplemental Figure 1. Clinical relevance of natural product methyltransferases (NPMTs) in brain disorders. (A) 4 Table summarizing characteristics of 11 NPMTs using data derived from the TCGA GBM and Rembrandt datasets for 5 relative expression levels and survival. In addition, published studies of the 11 NPMTs are summarized. (B) The 1 Jung et al. 6 expression levels of 10 NPMTs in glioblastoma versus non‐tumor brain are displayed in a heatmap, ranked by 7 significance and expression levels. *, p<0.05; **, p<0.01; ***, p<0.001. 8 2 Jung et al. 9 10 Supplemental Figure 2. Anatomical distribution of methyltransferase and metabolic signatures within 11 glioblastomas. The Ivy GAP dataset was downloaded and interrogated by histological structure for NNMT, NAMPT, 12 DNMT mRNA expression and selected gene expression signatures. The results are displayed on a heatmap. The 13 sample size of each histological region as indicated on the figure. 14 3 Jung et al. 15 16 Supplemental Figure 3. Altered expression of nicotinamide and nicotinate metabolism‐related enzymes in 17 glioblastoma. (A) Heatmap (fold change of expression) of whole 25 enzymes in the KEGG nicotinate and 18 nicotinamide metabolism gene set were analyzed in indicated glioblastoma expression datasets with Oncomine. 4 Jung et al. 19 Color bar intensity indicates percentile of fold change in glioblastoma relative to normal brain. (B) Nicotinamide and 20 nicotinate and methionine salvage pathways are displayed with the relative expression levels in glioblastoma 21 specimens in the TCGA GBM dataset indicated. 22 5 Jung et al. 23 24 Supplementary Figure 4. -

Microrna Hsa-Mir-1301-3P Regulates Human ADH6, ALDH5A1 and ALDH8A1 in the Ethanol-Acetaldehyde-Acetate Metabolic Pathway
Molecular Pharmacology Fast Forward. PublishedMOL # 1196on June93 4, 2020 as DOI: 10.1124/mol.120.119693 This article has not been copyedited and formatted. The final version may differ from this version. MicroRNA hsa-miR-1301-3p regulates human ADH6, ALDH5A1 and ALDH8A1 in the ethanol-acetaldehyde-acetate metabolic pathway Xubing Wang1*, Yanjie Zhao1*, Jiao Luo1, Lin Xu1, Xinmei Li1, Yuan Jin1, Chuanhai Li1, Meiyao Feng1, Ying Wang1, Jing Chen1, Yufei Hou1, Qianwen Zhao1, Jinquan Zhao1, Baitang Ning2, Yuxin Zheng1, and Dianke Yu1# Downloaded from 1School of Public Health, Qingdao University, Qingdao, China. 2National Center for Toxicological Research, US Food and Drug Administration. molpharm.aspetjournals.org # Corresponding Author: at ASPET Journals on September 25, 2021 E-mail address: [email protected] 1 Molecular Pharmacology Fast Forward. PublishedMOL # 1196on June93 4, 2020 as DOI: 10.1124/mol.120.119693 This article has not been copyedited and formatted. The final version may differ from this version. (1) Running Title: hsa-miR-1301-3p regulates ADH6, ALDH5A1, and ALDH8A1 (2) Corresponding author: Name: Dianke Yu Address: School of Public Health, Qingdao University, 38 Dengzhou Road, Qingdao, Shandong 266021, China. Telephone:86-13361480538 Downloaded from Email: [email protected] (3) Text pages: 30 Number of tables: 1 molpharm.aspetjournals.org Number of figures: 5 Number of references: 56 Number of words in the Abstract: 199 at ASPET Journals on September 25, 2021 Number of words in the Introduction: 709 Number of words -

906 Variations Among 27 Genes Encoding Cytochrome P450 (CYP) Enzymes and Aldehyde Dehydrogenases (Aldhs) in the Japanese Population
B.J Hum Jochimsen Genet et(2002) al.: Stetteria 47:419–444 hydrogenophila © Jpn Soc Hum Genet and Springer-Verlag4600/419 2002 ORIGINAL ARTICLE Susumu Saito · Aritoshi Iida · Akihiro Sekine Chie Ogawa · Saori Kawauchi · Shoko Higuchi Machi Ohno · Yusuke Nakamura 906 variations among 27 genes encoding cytochrome P450 (CYP) enzymes and aldehyde dehydrogenases (ALDHs) in the Japanese population Received: April 23, 2002 / Accepted: April 25, 2002 Abstract We screened DNAs from 48 Japanese individuals steroid hormones and xenobiotics, including various car- for single-nucleotide polymorphisms (SNPs) in genes en- cinogens and toxins (Denison and Whitlock 1995; Nelson et coding 13 cytochrome P450 (CYP) enzymes and 14 alde- al. 1996). CYP genes are classified into different families hyde dehydrogenases (ALDHs) by directly sequencing and subfamilies on the basis of sequence similarities. their entire genomic regions except for repetitive elements. Polymorphisms have been reported in CYP1A1, This approach identified 810 SNPs and 96 insertion/deletion CYP1A2, and CYP1B1 genes [Human Cytochrome polymorphisms among the 27 genes. Of the 810 SNPs, 229 P450 (CYP) Allele Nomenclature Committee, http:// were identified among the CYP genes and 581 in the ALDH www.imm.ki.se/CYPalleles/]. These three genes are genes; of the total, 48 SNPs were located in 5Ј flanking inducible by exposure to agents such as 2,3,7,8- regions, 619 in introns, 91 in exons, and 52 in 3Ј flanking tetrachlorodibenzo-p-dioxin (dioxin; Jaiswal et al. 1985, regions. These variants should contribute to studies 1987; Sutter et al. 1994) Two polymorphisms in the designed to investigate possible correlations between CYP1A1 gene, a T-to-C polymorphism in the 3Ј-noncoding genotypes and phenotypes of disease susceptibility or region and an A-to-G polymorphism (Ile462Val) in exon 7, responsiveness to drug therapy.