RNA Sequencing Reveals Upregulation of RUNX1-RUNX1T1 Gene Signatures in Clear Cell Renal Cell Carcinoma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Variations in Microrna-25 Expression Influence the Severity of Diabetic
BASIC RESEARCH www.jasn.org Variations in MicroRNA-25 Expression Influence the Severity of Diabetic Kidney Disease † † † Yunshuang Liu,* Hongzhi Li,* Jieting Liu,* Pengfei Han, Xuefeng Li, He Bai,* Chunlei Zhang,* Xuelian Sun,* Yanjie Teng,* Yufei Zhang,* Xiaohuan Yuan,* Yanhui Chu,* and Binghai Zhao* *Heilongjiang Key Laboratory of Anti-Fibrosis Biotherapy, Medical Research Center, Heilongjiang, People’s Republic of China; and †Clinical Laboratory of Hong Qi Hospital, Mudanjiang Medical University, Heilongjiang, People’s Republic of China ABSTRACT Diabetic nephropathy is characterized by persistent albuminuria, progressive decline in GFR, and second- ary hypertension. MicroRNAs are dysregulated in diabetic nephropathy, but identification of the specific microRNAs involved remains incomplete. Here, we show that the peripheral blood from patients with diabetes and the kidneys of animals with type 1 or 2 diabetes have low levels of microRNA-25 (miR-25) compared with those of their nondiabetic counterparts. Furthermore, treatment with high glucose decreased the expression of miR-25 in cultured kidney cells. In db/db mice, systemic administration of an miR-25 agomir repressed glomerular fibrosis and reduced high BP. Notably, knockdown of miR-25 in normal mice by systemic administration of an miR-25 antagomir resulted in increased proteinuria, extracellular matrix accumulation, podocyte foot process effacement, and hypertension with renin-angiotensin system activation. However, excessive miR-25 did not cause kidney dysfunction in wild-type mice. RNA sequencing showed the alteration of miR-25 target genes in antagomir-treated mice, including the Ras-related gene CDC42. In vitro,cotrans- fection with the miR-25 antagomir repressed luciferase activity from a reporter construct containing the CDC42 39 untranslated region. -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

The Oestrogen Receptor Alpha-Regulated Lncrna NEAT1 Is a Critical Modulator of Prostate Cancer
ARTICLE Received 5 Dec 2013 | Accepted 26 Sep 2014 | Published 21 Nov 2014 DOI: 10.1038/ncomms6383 OPEN The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer Dimple Chakravarty1,2, Andrea Sboner1,2,3, Sujit S. Nair4, Eugenia Giannopoulou5,6, Ruohan Li7, Sven Hennig8, Juan Miguel Mosquera1,2, Jonathan Pauwels1, Kyung Park1, Myriam Kossai1,2, Theresa Y. MacDonald1, Jacqueline Fontugne1,2, Nicholas Erho9, Ismael A. Vergara9, Mercedeh Ghadessi9, Elai Davicioni9, Robert B. Jenkins10, Nallasivam Palanisamy11,12, Zhengming Chen13, Shinichi Nakagawa14, Tetsuro Hirose15, Neil H. Bander16, Himisha Beltran1,2, Archa H. Fox7, Olivier Elemento2,3 & Mark A. Rubin1,2 The androgen receptor (AR) plays a central role in establishing an oncogenic cascade that drives prostate cancer progression. Some prostate cancers escape androgen dependence and are often associated with an aggressive phenotype. The oestrogen receptor alpha (ERa)is expressed in prostate cancers, independent of AR status. However, the role of ERa remains elusive. Using a combination of chromatin immunoprecipitation (ChIP) and RNA-sequencing data, we identified an ERa-specific non-coding transcriptome signature. Among putatively ERa-regulated intergenic long non-coding RNAs (lncRNAs), we identified nuclear enriched abundant transcript 1 (NEAT1) as the most significantly overexpressed lncRNA in prostate cancer. Analysis of two large clinical cohorts also revealed that NEAT1 expression is asso- ciated with prostate cancer progression. Prostate cancer cells expressing high levels of NEAT1 were recalcitrant to androgen or AR antagonists. Finally, we provide evidence that NEAT1 drives oncogenic growth by altering the epigenetic landscape of target gene promoters to favour transcription. 1 Department of Pathology and Laboratory Medicine, Weill Medical College of Cornell University, 413 East 69th Street, Room 1402, New York, New York 10021, USA. -

Inhibition of Single Minded 2 Gene Expression Mediates Tumor-Selective Apoptosis and Differentiation in Human Colon Cancer Cells
Inhibition of Single Minded 2 gene expression mediates tumor-selective apoptosis and differentiation in human colon cancer cells Mireille J. Aleman*†‡§, Maurice Phil DeYoung*†§¶, Matthew Tress*†, Patricia Keating*†, Gary W. Perryʈ, and Ramaswamy Narayanan*†** *Center for Molecular Biology and Biotechnology, Departments of †Biology and ‡Chemistry, and ¶Center for Complex System and Brain Sciences, Florida Atlantic University, 777 Glades Road, Boca Raton, FL 33431 Communicated by Herbert Weissbach, Florida Atlantic University, Boca Raton, FL, July 21, 2005 (received for review April 4, 2005) A Down’s syndrome associated gene, Single Minded 2 gene short bound AhR͞ARNT complex (12) and hence prevent carcinogen form (SIM2-s), is specifically expressed in colon tumors but not in metabolism, leading to cumulative DNA damage and cancer. the normal colon. Antisense inhibition of SIM2-s in a RKO-derived The growth arrest and DNA damage (GADD) family of genes colon carcinoma cell line causes growth inhibition, apoptosis, and was originally isolated from UV radiation-treated cells and subse- inhibition of tumor growth in a nude mouse tumoriginicity model. quently grouped according to their coordinate regulation by growth The mechanism of cell death in tumor cells is unclear. In the present arrest and DNA damage (13). The GADD family members include study, we investigated the pathways underlying apoptosis. Apo- GADD34,-45␣,-45,-45␥, and -153 (14, 15). These are stress- ptosis was seen in a tumor cell-specific manner in RKO cells but not response genes induced by both genotoxic and nongenotoxic in normal renal epithelial cells, despite inhibition of SIM2-s expres- stresses (16–18). GADD45␣ is the most extensively studied mem- sion in both of these cells by the antisense. -

Download Download
NEUROSCIENCE RESEARCH NOTES OPEN ACCESS | RESEARCH NOTES ISSN: 2576-828X Transcriptomic profiling of skeletal muscle from the Ts1Cje mouse model of Down syndrome suggests dysregulation of trisomic genes associated with neuromuscular junction signaling, oxidative stress and chronic inflammation Melody Pui Yee Leong 1,2, Usman Bala 2,3, Chai Ling Lim 2,3, Rozita Rosli 1,2, Pike-See Cheah 2,3 and King-Hwa Ling 1,2,* 1 Department of Biomedical Science, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia. 2 Genetics and Regenerative Medicine Research Centre (GRMRC), Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia. 3 Department of Human Anatomy, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia. * Correspondence: [email protected]; Tel.: +603-8947 2564 Received: 10 May 2018; Accepted: 21 August 2018; Published: 27 August 2018 Edited by: Azlina Ahmad Annuar (University of Malaya, Malaysia) Reviewed by: Ruth Chia (National Institute on Aging, National Institutes of Health, USA); Shelisa Tey (Institute of Neuropathology, RWTH University Hospital Aachen, Germany) DOI: https://doi.org/10.31117/neuroscirn.v1i1.12 Abstract: Ts1Cje is a mouse model of Down syndrome (DS) with partial triplication of chromosome 16, which encompasses a high number of human chromosome 21 (HSA21) orthologous genes. The mouse model exhibits muscle weakness resembling hypotonia in DS individuals. The effect of extra gene dosages on muscle weakness or hypotonia in Ts1Cje and DS individuals remains unknown. To identify molecular dysregulation of the skeletal muscle, we compared the transcriptomic signatures of soleus and extensor digitorum longus (EDL) muscles between the adult Ts1Cje and disomic littermates. -

The Nuclear Hormone Receptor Coactivator SRC-1 Is a Specific Target of P300 TSO-PANG YAO, GREGORY Ku, NAIDONG ZHOU, RALPH SCULLY, and DAVID M
Proc. Natl. Acad. Sci. USA Vol. 93, pp. 10626-10631, October 1996 Biochemistry The nuclear hormone receptor coactivator SRC-1 is a specific target of p300 TSO-PANG YAO, GREGORY Ku, NAIDONG ZHOU, RALPH SCULLY, AND DAVID M. LIvINGSTON* Dana-Farber Cancer Institute and Harvard Medical School, 44 Binney Street, Boston, MA 02115 Contributed by David M. Livingston, June 17, 1996 ABSTRACT p300 and its family member, CREB-binding derlying ligand activation of nuclear receptor transcription protein (CBP), function as key transcriptional coactivators by activity. virtue of their interaction with the activated forms of certain Specifically, it has been shown that, upon ligand binding, a transcription factors. In a search for additional cellular specific set of proteins are recruited to the hormone bound targets of p300/CBP, a protein-protein cloning strategy, (i.e., activated) receptor (11-13). It was hypothesized that surprisingly identified SRC-1, a coactivator involved in nu- these multiprotein complexes assemble and become transcrip- clear hormone receptor transcriptional activity, as a p300/ tionally active in response to hormone binding. Identifying and CBP interactive protein. p300 and SRC-1 interact, specifically, understanding how individual components of these complexes in vitro and they also form complexes in vivo. Moreover, we function are part of the key to understand how nuclear show that SRC-1 encodes a new member of the basic helix- receptors modulate transcription. Recently, one of the nuclear loop-helix-PAS domain family and that it physically interacts receptor-binding proteins, steroid receptor coactivator-1 with the retinoic acid receptor in response to hormone bind- (SRC-1), was identified and cloned. -

The Role of Npas4 in Neuroprotection and Neurogenesis
!!"$ &#!"#!%%! &#! $$ ! !( %$$$&%%!#%#! !%!#!!$!"( "#% % ! %$ '!&%! !!! !! $ '#$%(! # ,01;4 >5>=C4=CB 42;0A0C8>= 2:=>F;4364<4=CB 1BCA02C 70?C4A "=CA>3D2C8>= *4E84F(0?4A *46D;0C8>=>5'?0B4G?A4BB8>=1H>C74A<4270=8B<B -*',*'("#&(&&,(+ 0(*++#'& (+ 0(*++#'&#&*+('&+,'# *&,,1(+' +,*++ 338C8>=0;5D=2C8>=B>5'?0B 0#,,#'&#&"##,#'&"'%'+,+#+ -*'!&+#+ &82A>*'B #'!&+#+' %# + %# +#&&-*'!&+#+ (A>942CA0C8>=0;40=3A4B40A2708<B 70?C4A *4B40A27?0?4A 70?C4A *4B40A27?0?4A 70?C4A 8BC8=2C8E4?A>58;4>58B><8*4G?A4BB8>=0=3=>E4;<82A>*'B3DA8=6=4DA0; 38554A4=C80C8>=>5<>DB44<1AH>=82BC4<24;;B "=CA>3D2C8>= *4BD;CB0=338B2DBB8>= .*.#/' +%$$ +)-&#&!, -&&' #+'%#+)-&+ &,# #,#'&' &'.$%# + >=2;DB8>= 70?C4A >=2;D38=6*4<0A:B (A>942CB86=85820=24 *42><<4=30C8>=B5>A5DAC74AF>A: *454A4=24 42;0A0C8>= 5FGQUMPIAMLR?GLQLMK?RCPG?JUFGAFF?Q@CCL?AACNRCBDMPRFC?U?PBMD?LWMRFCPBCEPCC MPBGNJMK?GL?LWSLGTCPQGRWMPMRFCPRCPRG?PWGLQRGRSRGML?LB RMRFC@CQRMDKWILMUJCBEC ?LB@CJGCD AMLR?GLQLMK?RCPG?JNPCTGMSQJWNS@JGQFCBMPUPGRRCL@W?LMRFCP NCPQML CVACNR UFCPCBSCPCDCPCLACF?Q@CCLK?BCGLRFCRCVR *EGTCAMLQCLRRMRFGQAMNWMDKWRFCQGQ UFCLBCNMQGRCBGLRFC6LGTCPQGRW-G@P?PW @CGLE K?BC?T?GJ?@JCDMPJM?L?LBNFMRMAMNWGLE QS@HCARRMRFCNPMTGQGMLQMDRFC$MNWPGEFR"AR *?JQM EGTCNCPKGQQGMLDMPRFCBGEGR?JTCPQGMLMDKWRFCQGQRM@CK?BC?T?GJ?@JCMLRFCUC@ TG?RFC6LGTCPQGRWmQBGEGR?JPCQC?PAFPCNMQGRMPW RFC-G@P?PWA?R?JMESC?LB?JQMRFPMSEFUC@ QC?PAFCLEGLCQ SLJCQQNCPKGQQGMLF?Q@CCLEP?LRCB@WRFC6LGTCPQGRWRMPCQRPGAR?AACQQDMP? NCPGMBMDRGKC 4GELCB %?RC 2:=>F;4364<4=CB *UMSJBJGICRMR?ICRFGQMNNMPRSLGRWRMCVNPCQQKWSRKMQREP?RGRSBCRMK?LWGLBGTGBS?JQUFM -

Ohnologs in the Human Genome Are Dosage Balanced and Frequently Associated with Disease
Ohnologs in the human genome are dosage balanced and frequently associated with disease Takashi Makino1 and Aoife McLysaght2 Smurfit Institute of Genetics, University of Dublin, Trinity College, Dublin 2, Ireland Edited by Michael Freeling, University of California, Berkeley, CA, and approved April 9, 2010 (received for review December 21, 2009) About 30% of protein-coding genes in the human genome are been duplicated by WGD, subsequent loss of individual genes related through two whole genome duplication (WGD) events. would result in a dosage imbalance due to insufficient gene Although WGD is often credited with great evolutionary impor- product, thus leading to biased retention of dosage-balanced tance, the processes governing the retention of these genes and ohnologs. In fact, evidence for preferential retention of dosage- their biological significance remain unclear. One increasingly pop- balanced genes after WGD is accumulating (4, 7, 11–20). Copy ular hypothesis is that dosage balance constraints are a major number variation [copy number polymorphism (CNV)] describes determinant of duplicate gene retention. We test this hypothesis population level polymorphism of small segmental duplications and show that WGD-duplicated genes (ohnologs) have rarely and is known to directly correlate with gene expression levels (21– experienced subsequent small-scale duplication (SSD) and are also 24). Thus, CNV of dosage-balanced genes is also expected to be refractory to copy number variation (CNV) in human populations deleterious. This model predicts that retained ohnologs should be and are thus likely to be sensitive to relative quantities (i.e., they are enriched for dosage-balanced genes that are resistant to sub- dosage-balanced). -

The Molecular Pathogenesis of Down Syndrome-Associated Diseases And
Clinical and Medical Investigations Editorial ISSN: 2398-5763 The molecular pathogenesis of down syndrome-associated diseases and the promising potential therapeutic targets Mohammed Rachidi Molecular Genetics of Human Diseases (MGHD), French Polynesia and University Paris 7 Denis Diderot, Paris, France Trisomy 21, the most common origin of Down syndrome, is an development [7]. Colon carcinoma cell lines selected for increased extra copy of chromosome 21 in all cells generated by a chromosomal metastatic potential in nude mice express more TIAM1 protein non-disjunction during meiosis. The increased transcription of than their parental line [8,9]. This indicate that TIAM1 may have a the oncogenes and tumor suppressor genes located on the human role in the progression and metastasis of colon carcinomas and that chromosome 21 determines transcriptional alterations of several TIAM1 regulates cell adhesion, migration and apoptosis in colon molecular pathways involved in the Down syndrome associated tumor cells. RNA interference (RNAi) study examined the effect of diseases related to cell cycle alteration, leukemia, tumors and cancer. the inhibition of TIAM1 expression on proliferation and metastasis The elucidation of the molecular mechanisms involved in the Down and it has been found that the silencing of TIAM1 resulted in the syndrome-associated diseases could promise potential therapeutic effective inhibition of in vitro cell growth and of the invasive ability of targets. colorectal cancer cells. This suggest that TIAM1 plays a causal role in the metastasis of colorectal cancer and that RNAi-mediated silencing Trisomy 21 or Down syndrome determined by the triplication of of TIAM1 may provide an opportunity to develop a new treatment human chromosome 21, is the most frequent genetic disorder with strategy for colorectal cancer [10]. -
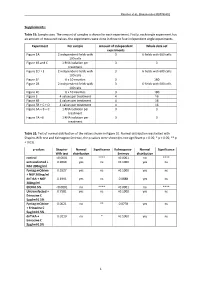
1 Supplements
Rascher et al., Biomolecules MS#748802 Supplements: Table S1: Sample sizes. The amount of samples is shown for each experiment. Firstly, each single experiment has an amount of measured values, the experiments were done in three to four independent single experiments. Experiment Per sample Amount of independent Whole data set experiments Figure 1A 2 independent fields with 3 6 fields with 600 cells 100 cells Figure 1B and C 1 RNA isolation per 3 3 treatment Figure 1D + E 2 independent fields with 3 6 fields with 600 cells 100 cells Figure 1F 6 x 10 neurites 3 180 Figure 2B 2 independent fields with 3 6 fields with 600 cells 100 cells Figure 2C 6 x 10 neurites 3 180 Figure 3 4 values per treatment 4 16 Figure 4B 4 values per treatment 4 16 Figure 5B + C + D 4 values per treatment 4 16 Figure 6A + B + C 1 RNA isolation per 3 3 treatment Figure 7A +B 1 RNA isolation per 3 3 treatment Table S2: Test of normal distribution of the values shown in Figure 1E. Normal distribution was tested with Shapiro-Wilk test and Kolmogrov-Smirnov, the p-values were shown (ns not significant p > 0.05; * p < 0.05; ** p < 0.01). p-values Shapiro- Normal Significance Kolmogorov- Normal Significance Wilk test distribution Smirnov distribution control <0.0001 no **** <0.0001 no **** untransfected + 0.9069 yes ns >0.1000 yes ns NGF 200ng/ml Fyntag mCitrine 0.5527 yes ns >0.1000 yes ns + NGF 200ng/ml dnTrkA + NGF 0.1991 yes ns 0.0888 yes ns 200ng/ml EtOH 0.5% <0.0001 no **** <0.0001 no **** Untransfected + 0.7581 yes ns >0.1000 yes ns Erinacine C 5µg/ml 0.5% Fyntag mCitrine 0.0021 no ** 0.0778 yes ns + Erinacine C 5µg/ml 0.5% dnTrkA + 0.0110 no * >0.1000 yes ns Erinacine C 5µg/ml 0.5% 1 Rascher et al., Biomolecules MS#748802 Table S3: Test of normal distribution of the values shown in Figure 1F. -

Frequency and Clinical Characteristics of Intrachromosomal Amplification of Chromosome 21 in Korean Childhood B-Lineage Acute Lymphoblastic Leukemia
Original Article Diagnostic Genetics Ann Lab Med 2016;36:475-480 http://dx.doi.org/10.3343/alm.2016.36.5.475 ISSN 2234-3806 • eISSN 2234-3814 Frequency and Clinical Characteristics of Intrachromosomal Amplification of Chromosome 21 in Korean Childhood B-lineage Acute Lymphoblastic Leukemia Jieun Kim, M.D.1, Chuhl Joo Lyu, M.D.2, Saeam Shin, M.D.3, Seung-Tae Lee, M.D.1, and Jong Rak Choi, M.D.1 Department of Laboratory Medicine1, Yonsei University College of Medicine, Seoul; Department of Pediatrics2, Yonsei Cancer Research Center, Yonsei University College of Medicine, Seoul; Department of Laboratory Medicine3, Hallym University College of Medicine, Kangnam Sacred Heart Hospital, Seoul, Korea Background: Intrachromosomal amplification of chromosome 21 (iAMP21) is known to Received: December 1, 2015 be associated with poor prognosis in B-cell ALL (B-ALL). To determine the frequency and Revision received: March 30, 2016 Accepted: May 11, 2016 clinical characteristics of iAMP21 in Korean B-ALL patients, we performed FISH and mul- tiplex ligation-dependent probe amplification (MLPA) analyses. Corresponding author: Seung-Tae Lee Department of Laboratory Medicine, Yonsei Methods: A total of 102 childhood B-ALL patients were screened with ETV6-RUNX1 FISH University College of Medicine, 50-1 probes (Abbott Molecular, USA). The presence of an iAMP21 was confirmed by using Yonsei-ro, Seodaemun-gu, Seoul 03722, Korea MLPA P327 iAMP21-ERG probemix (MRC Holland, The Netherlands). Tel: +82-2-2228-2450 Results: iAMP21 was detected in one of the screened B-ALL patients (1/102 patients, Fax: +82-2-364-1583 E-mail: [email protected] 1.0%) who presented the ALL immunophenotype and complex karyotype at initial diagno- sis. -

Canonical WNT Signalling Determines Lineage Specificity in Wilms Tumour
Oncogene (2009) 28, 1063–1075 & 2009 Macmillan Publishers Limited All rights reserved 0950-9232/09 $32.00 www.nature.com/onc ORIGINAL ARTICLE Canonical WNT signalling determines lineage specificity in Wilms tumour R Fukuzawa, MR Anaka, RJ Weeks, IM Morison and AE Reeve Cancer Genetics Laboratory, Department of Biochemistry, University of Otago, Dunedin, New Zealand Wilms tumours (WTs) have two distinct types of histology to translocation of the protein to the nucleus (Clevers, with or without ectopic mesenchymal elements, suggesting 2006). Nuclear accumulation of b-catenin is therefore a that WTs arise from either the mesenchymal or epithelial surrogate marker for activation of the WNT-signalling nephrogenic lineages. Regardless of the presence or pathway in many types of cancers (Clevers, 2006). absence of CTNNB1 mutations, nuclear accumulation of Wilms tumour (WT) is an embryonal tumour that b-catenin is often observed in WTs with ectopic mesench- recapitulates kidney development. WT typically has a ymal elements. Here, we addressed the relationship triphasic histology, which consists of varying propor- between the WNT-signalling pathway and lineage in tions of blastemal, epithelial and stromal elements WTs by examining CTNNB1 and WT1 mutations, (Beckwith et al., 1990). The stromal tissues occasionally nuclear accumulation of b-catenin, tumour histology and contain ectopic mesenchymal elements such as rhabdo- gene expression profiles. In addition, we screened for myoblasts, chondrocytes and adipocytes. Remarkably, mutations in WTX, which has been proposed to be a the presence or absence of ectopic mesenchymal negative regulator of the canonical WNT-signalling elements in WT histology is associated with specific pathway. Unsupervised clustering analysis identified two precursor lesions, intralobar and perilobar nephrogenic classes of tumours: mesenchymal lineage WNT-dependent rests (ILNR and PLNR), respectively (Beckwith et al., tumours, and epithelial lineage WNT-independent tu- 1990).