Allele-Specific Expression Links Diverse Cis-Regulatory Mutations to Recurrent Gene Dysregulation in High-Risk Neuroblastoma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Plekhg5-Regulated Autophagy of Synaptic Vesicles Reveals a Pathogenic Mechanism in Motoneuron Disease
Thomas Jefferson University Jefferson Digital Commons Department of Medicine Faculty Papers Department of Medicine 12-1-2017 Plekhg5-regulated autophagy of synaptic vesicles reveals a pathogenic mechanism in motoneuron disease. Patrick Lüningschrör University Hospital Würzburg; University of Bielefeld Beyenech Binotti Max Planck Institute for Biophysical Chemistry, Göttingen Benjamin Dombert University Hospital Würzburg Follow this and additional works at: https://jdc.jefferson.edu/medfp Peter Heimann Univ Persityart of of the Bielef Neureldology Commons AngelLet usPerez-Lar knowa how access to this document benefits ouy University of Bielefeld Recommended Citation Lüningschrör, Patrick; Binotti, Beyenech; Dombert, Benjamin; Heimann, Peter; Perez-Lara, Angel; See next page for additional authors Slotta, Carsten; Thau-Habermann, Nadine; von Collenberg, Cora R.; Karl, Franziska; Damme, Markus; Horowitz, Arie; Maystadt, Isabelle; Füchtbauer, Annette; Füchtbauer, Ernst-Martin; Jablonka, Sibylle; Blum, Robert; Üçeyler, Nurcan; Petri, Susanne; Kaltschmidt, Barbara; Jahn, Reinhard; Kaltschmidt, Christian; and Sendtner, Michael, "Plekhg5-regulated autophagy of synaptic vesicles reveals a pathogenic mechanism in motoneuron disease." (2017). Department of Medicine Faculty Papers. Paper 220. https://jdc.jefferson.edu/medfp/220 This Article is brought to you for free and open access by the Jefferson Digital Commons. The Jefferson Digital Commons is a service of Thomas Jefferson University's Center for Teaching and Learning (CTL). The Commons is a showcase for Jefferson books and journals, peer-reviewed scholarly publications, unique historical collections from the University archives, and teaching tools. The Jefferson Digital Commons allows researchers and interested readers anywhere in the world to learn about and keep up to date with Jefferson scholarship. This article has been accepted for inclusion in Department of Medicine Faculty Papers by an authorized administrator of the Jefferson Digital Commons. -

Genetic Modifiers of Hereditary Neuromuscular Disorders
cells Article Genetic Modifiers of Hereditary Neuromuscular Disorders and Cardiomyopathy Sholeh Bazrafshan 1, Hani Kushlaf 2 , Mashhood Kakroo 1, John Quinlan 2, Richard C. Becker 1 and Sakthivel Sadayappan 1,* 1 Heart, Lung and Vascular Institute, Division of Cardiovascular Health and Disease, Department of Internal Medicine, University of Cincinnati College of Medicine, Cincinnati, OH 45267, USA; [email protected] (S.B.); [email protected] (M.K.); [email protected] (R.C.B.) 2 Department of Neurology and Rehabilitation Medicine, Neuromuscular Center, University of Cincinnati Gardner Neuroscience Institute, University of Cincinnati College of Medicine, Cincinnati, OH 45267, USA; [email protected] (H.K.); [email protected] (J.Q.) * Correspondence: [email protected]; Tel.: +1-513-558-7498 Abstract: Novel genetic variants exist in patients with hereditary neuromuscular disorders (NMD), including muscular dystrophy. These patients also develop cardiac manifestations. However, the association between these gene variants and cardiac abnormalities is understudied. To determine genetic modifiers and features of cardiac disease in NMD patients, we have reviewed electronic medical records of 651 patients referred to the Muscular Dystrophy Association Care Center at the University of Cincinnati and characterized the clinical phenotype of 14 patients correlating with their next-generation sequencing data. The data were retrieved from the electronic medical records of the 14 patients included in the current study and comprised neurologic and cardiac phenotype and genetic reports which included comparative genomic hybridization array and NGS. Novel associations were uncovered in the following eight patients diagnosed with Limb-girdle Muscular Dystrophy, Bethlem Myopathy, Necrotizing Myopathy, Charcot-Marie-Tooth Disease, Peripheral Citation: Bazrafshan, S.; Kushlaf, H.; Kakroo, M.; Quinlan, J.; Becker, R.C.; Polyneuropathy, and Valosin-containing Protein-related Myopathy. -

Biological Role and Disease Impact of Copy Number Variation in Complex Disease
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2014 Biological Role and Disease Impact of Copy Number Variation in Complex Disease Joseph Glessner University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, and the Genetics Commons Recommended Citation Glessner, Joseph, "Biological Role and Disease Impact of Copy Number Variation in Complex Disease" (2014). Publicly Accessible Penn Dissertations. 1286. https://repository.upenn.edu/edissertations/1286 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/1286 For more information, please contact [email protected]. Biological Role and Disease Impact of Copy Number Variation in Complex Disease Abstract In the human genome, DNA variants give rise to a variety of complex phenotypes. Ranging from single base mutations to copy number variations (CNVs), many of these variants are neutral in selection and disease etiology, making difficult the detection of true common orar r e frequency disease-causing mutations. However, allele frequency comparisons in cases, controls, and families may reveal disease associations. Single nucleotide polymorphism (SNP) arrays and exome sequencing are popular assays for genome-wide variant identification. oT limit bias between samples, uniform testing is crucial, including standardized platform versions and sample processing. Bases occupy single points while copy variants occupy segments. -

Genetics of Distal Hereditary Motor Neuropathies
GENETICSOFDISTALHEREDITARY MOTOR NEUROPATHIES By alexander peter drew A thesis submitted for the Degree of Doctor of Philosophy Supervised by Professor Garth A. Nicholson Dr. Ian P. Blair Faculty of Medicine University of Sydney 2012 statement No part of the work described in this thesis has been submitted in fulfilment of the requirements for any other academic degree or qualification. Except where due acknowledgement has been made, all experimental work was performed by the author. Alexander Peter Drew CONTENTS acknowledgements ............................. i summary .................................... ii list of figures ................................ v list of tables ................................ viii acronyms and abbreviations ..................... xi publications ................................. xiv 1 literature review ........................... 1 1.1 Molecular genetics and mechanisms of disease in Distal Hereditary Motor Neuropathies . .1 1.1.1 Small heat shock protein family . .2 1.1.2 Dynactin 1 (DCTN1).....................9 1.1.3 Immunoglobulin mu binding protein 2 gene (IGHMBP2) 11 1.1.4 Senataxin (SETX)....................... 14 1.1.5 Glycyl-tRNA synthase (GARS)............... 16 1.1.6 Berardinelli-Seip congenital lipodystrophy 2 (SEIPIN) gene (BSCL2)......................... 18 1.1.7 ATPase, Cu2+-transporting, alpha polypeptide gene (ATP7A) 20 1.1.8 Pleckstrin homology domain-containing protein, G5 gene (PLEKHG5)........................... 21 1.1.9 Transient receptor potential cation channel, V4 gene (TRPV4) 22 1.1.10 DYNC1H1 ........................... 23 1.1.11 Clinical variability in dHMN . 24 1.1.12 Common disease mechanisms in dHMN . 29 2 general materials and methods ................. 32 2.1 General materials and reagents . 32 2.1.1 Reagents and Enzymes . 32 2.1.2 Equipment . 33 2.1.3 Plasticware . 33 2.2 Study participants . 34 2.3 DNA methods . -

Genetic and Functional Investigation of Inherited Neuropathies
GENETIC AND FUNCTIONAL INVESTIGATION OF INHERITED NEUROPATHIES Ellen Cottenie MRC Centre for Neuromuscular Diseases, UCL Institute of Neurology Supervisors: Professor Mary M. Reilly, Professor Henry Houlden and Professor Mike Hanna Thesis submitted for the degree of Doctor of Philosophy University College London 2015 1 Declaration I, Ellen Cottenie, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. 2 Abstract With the discovery of next generation sequencing techniques the landscape of pathogenic gene discovery has shifted drastically over the last ten years. For the purpose of this thesis, focus was applied on finding genetic causes of inherited neuropathies, mainly Charcot-Marie-Tooth disease, by using both old and new genetic techniques and the accompanying functional investigations to prove the pathogenicity of these variants. Mutations in ATPase 6, the first mitochondrially encoded gene responsible for an isolated neuropathy, were found in five families with CMT2 by a traditional Sanger sequencing approach. The same approach was used to expand the phenotype associated with FIG4 mutations, known as CMT4J. Compound heterozygous mutations were found in a patient with a proximal and asymmetric weakness and rapid deterioration of strength in a single limb, mimicking CIDP. Several appropriate cohorts were screened for mutations in candidate genes with the traditional Sanger sequencing approach; however, no new pathogenic genes were found. In the case of the HINT1 gene, the originally stated frequency of 11% could not be replicated and a founder effect was suggested, underlying the importance of considering the ethnic background of a patient when screening for mutations in neuropathy-related genes. -
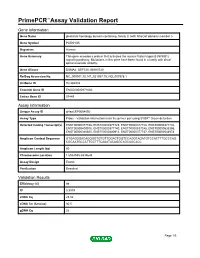
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name pleckstrin homology domain containing, family G (with RhoGef domain) member 5 Gene Symbol PLEKHG5 Organism Human Gene Summary This gene encodes a protein that activates the nuclear factor kappa B (NFKB1) signaling pathway. Multations in this gene have been found in a family with distal spinal muscular atrophy. Gene Aliases DSMA4, GEF720, KIAA0720 RefSeq Accession No. NC_000001.10, NT_021937.19, NG_007978.1 UniGene ID Hs.284232 Ensembl Gene ID ENSG00000171680 Entrez Gene ID 57449 Assay Information Unique Assay ID qHsaCEP0054092 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000377748, ENST00000377725, ENST00000377728, ENST00000377732, ENST00000400915, ENST00000377740, ENST00000537245, ENST00000535355, ENST00000340850, ENST00000400913, ENST00000377737, ENST00000544978 Amplicon Context Sequence GTGAGGGACAGGGGTGTGTTGGACTGGTCCAGGTAGATGTCCACTTTGCCCAG CGCAATGCCCTTCCTTTCAAATACAGGCAGCAGCACC Amplicon Length (bp) 60 Chromosome Location 1:6534555-6534644 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 98 R2 0.9979 cDNA Cq 25.02 cDNA Tm (Celsius) 80.5 gDNA Cq 26 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report PLEKHG5, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

A Yeast-Based Model for Hereditary Motor and Sensory Neuropathies: a Simple System for Complex, Heterogeneous Diseases
International Journal of Molecular Sciences Review A Yeast-Based Model for Hereditary Motor and Sensory Neuropathies: A Simple System for Complex, Heterogeneous Diseases Weronika Rzepnikowska 1, Joanna Kaminska 2 , Dagmara Kabzi ´nska 1 , Katarzyna Bini˛eda 1 and Andrzej Kocha ´nski 1,* 1 Neuromuscular Unit, Mossakowski Medical Research Centre Polish Academy of Sciences, 02-106 Warsaw, Poland; [email protected] (W.R.); [email protected] (D.K.); [email protected] (K.B.) 2 Institute of Biochemistry and Biophysics Polish Academy of Sciences, 02-106 Warsaw, Poland; [email protected] * Correspondence: [email protected] Received: 19 May 2020; Accepted: 15 June 2020; Published: 16 June 2020 Abstract: Charcot–Marie–Tooth (CMT) disease encompasses a group of rare disorders that are characterized by similar clinical manifestations and a high genetic heterogeneity. Such excessive diversity presents many problems. Firstly, it makes a proper genetic diagnosis much more difficult and, even when using the most advanced tools, does not guarantee that the cause of the disease will be revealed. Secondly, the molecular mechanisms underlying the observed symptoms are extremely diverse and are probably different for most of the disease subtypes. Finally, there is no possibility of finding one efficient cure for all, or even the majority of CMT diseases. Every subtype of CMT needs an individual approach backed up by its own research field. Thus, it is little surprise that our knowledge of CMT disease as a whole is selective and therapeutic approaches are limited. There is an urgent need to develop new CMT models to fill the gaps. -

Research and Diagnostic Validity of Whole Exome Sequencing in Neuromuscular Disease
Research and Diagnostic Validity of Whole Exome Sequencing in Neuromuscular Disease Linran Zhou A thesis submitted to the faculty at the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the Senior Honors Thesis in the Department of Biology in the College of Arts and Sciences. Chapel Hill 2015 Approved: Thesis Advisor Reader Reader ABSTRACT Linran Zhou: Research and Diagnostic Validity of Whole Exome Sequencing in Neuromuscular Disease (Under the direction of Jonathan S. Berg) Neuromuscular diseases (NMDs) are a broad group of inherited genetic disorders with a total global prevalence exceeding 1 in 3,000. Affecting the muscle and peripheral nervous system, NMDs result in significant disability and despite increased understanding of their molecular basis, the cause is unknown in the majority of patients. In an NCGENES patient cohort, we examined the diagnostic yield of whole exome sequencing (WES) for variants in genes currently associated with NMDs, neuropathies, and myopathies, and we evaluated the literature for known information about these genes. We analyzed the variants from WES in genes not on the current diagnostic lists to search for novel, rare, deleterious variants in possible disease candidate genes and identify patient candidates for further molecular analysis. Based on literature and clinical reports, we also evaluated the construction of the current diagnostic lists, expanding the myopathy and neuropathy lists by over 80 genes each. The lists were curated so that NMD list encompassed all myopathy and neuropathy genes, while the myopathy and neuropathy lists remained as unique as possible. Based on our overall analysis, we constructed a schematic to aid clinicians in deciding when to apply WES for patients with a neuromuscular condition of suspected genetic etiology. -

Analyzing the Mirna-Gene Networks to Mine the Important Mirnas Under Skin of Human and Mouse
Hindawi Publishing Corporation BioMed Research International Volume 2016, Article ID 5469371, 9 pages http://dx.doi.org/10.1155/2016/5469371 Research Article Analyzing the miRNA-Gene Networks to Mine the Important miRNAs under Skin of Human and Mouse Jianghong Wu,1,2,3,4,5 Husile Gong,1,2 Yongsheng Bai,5,6 and Wenguang Zhang1 1 College of Animal Science, Inner Mongolia Agricultural University, Hohhot 010018, China 2Inner Mongolia Academy of Agricultural & Animal Husbandry Sciences, Hohhot 010031, China 3Inner Mongolia Prataculture Research Center, Chinese Academy of Science, Hohhot 010031, China 4State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming 650223, China 5Department of Biology, Indiana State University, Terre Haute, IN 47809, USA 6The Center for Genomic Advocacy, Indiana State University, Terre Haute, IN 47809, USA Correspondence should be addressed to Yongsheng Bai; [email protected] and Wenguang Zhang; [email protected] Received 11 April 2016; Revised 15 July 2016; Accepted 27 July 2016 Academic Editor: Nicola Cirillo Copyright © 2016 Jianghong Wu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Genetic networks provide new mechanistic insights into the diversity of species morphology. In this study, we have integrated the MGI, GEO, and miRNA database to analyze the genetic regulatory networks under morphology difference of integument of humans and mice. We found that the gene expression network in the skin is highly divergent between human and mouse. -

Rnd3 in Cancer: a Review of the Evidence for Tumor Promoter Or Suppressor Lisa Paysan1,2,Leo� Piquet1,2,Fred� Eric� Saltel1,2, and Violaine Moreau1,2
Published OnlineFirst August 23, 2016; DOI: 10.1158/1541-7786.MCR-16-0164 Review Molecular Cancer Research Rnd3 in Cancer: A Review of the Evidence for Tumor Promoter or Suppressor Lisa Paysan1,2,Leo Piquet1,2,Fred eric Saltel1,2, and Violaine Moreau1,2 Abstract Rho-GTPases are members of the Ras superfamily of small squamous cell carcinoma. Although there appears to be no GTPases and are general modulators of important cellular general consensus about Rnd3 expressionincancersasthis processes in tumor biology such as migration and proliferation. protein is differently altered according to the tumor context, Among these proteins, Rnd3/RhoE, an atypical Rho-GTPase these alterations overwhelmingly favor a protumorigenic role. devoid of GTP hydrolytic activity, has recently been studied for Thus, depending on the tumor type, it may behave either as a its putative role in tumorigenesis. Indeed, Rnd3 is implicated in tumor suppressor or as a tumor promoter. Importantly, the processes, such as proliferation and migration, whose deregu- deregulation of Rnd3, in most cases, is linked to patient poor lation is linked to cancer development and metastasis. The aim outcome. of this review is to provide an overview of the data surrounding Rnd3 deregulation in cancers, its origin, and consequences. Implications: Rnd3 has prognostic marker potential as exem- Presented here is a comprehensive account of the expression plified in lung cancers and Rnd3 or Rnd3-associated sig- status and biological output obtained in prostate, liver, stom- naling pathways may represent a new putative therapeutic ach, colon, lung, and brain cancers as well as in melanoma and target. Mol Cancer Res; 14(11); 1033–44. -

POSITIVE: Pathogenic Mutation Detected
SAMPLE REPORT - 07/24/2019 Ordered By Contact ID:1524667 Org ID:1 Patient Name: 9570_Test, ABC Physician: Unknown, Unknown, MD Accession #: 00-125751 Specimen #: Ph:N/A Fx:N/A AP2 Order #: 705073 Specimen: Blood EDTA Client: Ambry (Purple top) Birthdate: 01/01/1976 Age: 43y 6m CA US Gender: M Additional Authorized Recipient: MRN #: N/A Collected: N/A Thai, Julia Ph:(949) 900-5500 Fx:(949) 900-5501 Indication: Internal Testing Received: 07/15/2019 Ethnicity: Caucasian NeuropathySelect: Analyses of 81 Genes Associated with Hereditary Neuropathy RESULTS TTR Pathogenic Mutation: p.V50M SUMMARY POSITIVE: Pathogenic Mutation Detected INTERPRETATION This individual is heterozygous for the p.V50M pathogenic mutation in the TTR gene. This result is consistent with a diagnosis of transthyretin (TTR) amyloidosis. The expression and severity of disease for this individual cannot be predicted. Genetic testing for pathogenic mutations in family members can be helpful in identifying at-risk individuals. Genetic counseling is a recommended option for all individuals undergoing genetic testing. No additional pathogenic mutations, variants of unknown significance, or gross deletions or duplications were detected. Genes Analyzed (81 total): ATL1, DNAJB2, DST, FAM134B, FGD4, FIG4, FUS, GAN, GDAP1, GJB1, HINT1, HSPB1, IGHMBP2, IKBKAP, KIF1A, LMNA, LRSAM1, MFN2, MPZ, MTMR2, NDRG1, NEFL, NGF, NTRK1, OPTN, PLEKHG5, PMP22, PRDM12, PRX, REEP1, SBF2, SCN9A, SH3TC2, SIGMAR1, SLC25A46, SLC52A2, SLC52A3, SPG11, TTR, VRK1 and WNK1 (sequencing and deletion/duplication); AARS, AIFM1, APOA1, ATL3, ATP7A, BICD2, BSCL2, CHCHD10, DCTN1, DNM2, DNMT1, DYNC1H1, EGR2, FBXO38, GARS, GNB4, GSN, HARS, HSPB8, INF2, LITAF, MARS, MORC2, NEFH, PDK3, PRPS1, RAB7A, SCN10A, SCN11A, SETX, SLC5A7, SPTLC1, SPTLC2, TARDBP, TFG, TRPV4, UBA1, VAPB, VCP and YARS (sequencing only).