POSITIVE: Pathogenic Mutation Detected
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Entrez Symbols Name Termid Termdesc 117553 Uba3,Ube1c
Entrez Symbols Name TermID TermDesc 117553 Uba3,Ube1c ubiquitin-like modifier activating enzyme 3 GO:0016881 acid-amino acid ligase activity 299002 G2e3,RGD1310263 G2/M-phase specific E3 ubiquitin ligase GO:0016881 acid-amino acid ligase activity 303614 RGD1310067,Smurf2 SMAD specific E3 ubiquitin protein ligase 2 GO:0016881 acid-amino acid ligase activity 308669 Herc2 hect domain and RLD 2 GO:0016881 acid-amino acid ligase activity 309331 Uhrf2 ubiquitin-like with PHD and ring finger domains 2 GO:0016881 acid-amino acid ligase activity 316395 Hecw2 HECT, C2 and WW domain containing E3 ubiquitin protein ligase 2 GO:0016881 acid-amino acid ligase activity 361866 Hace1 HECT domain and ankyrin repeat containing, E3 ubiquitin protein ligase 1 GO:0016881 acid-amino acid ligase activity 117029 Ccr5,Ckr5,Cmkbr5 chemokine (C-C motif) receptor 5 GO:0003779 actin binding 117538 Waspip,Wip,Wipf1 WAS/WASL interacting protein family, member 1 GO:0003779 actin binding 117557 TM30nm,Tpm3,Tpm5 tropomyosin 3, gamma GO:0003779 actin binding 24779 MGC93554,Slc4a1 solute carrier family 4 (anion exchanger), member 1 GO:0003779 actin binding 24851 Alpha-tm,Tma2,Tmsa,Tpm1 tropomyosin 1, alpha GO:0003779 actin binding 25132 Myo5b,Myr6 myosin Vb GO:0003779 actin binding 25152 Map1a,Mtap1a microtubule-associated protein 1A GO:0003779 actin binding 25230 Add3 adducin 3 (gamma) GO:0003779 actin binding 25386 AQP-2,Aqp2,MGC156502,aquaporin-2aquaporin 2 (collecting duct) GO:0003779 actin binding 25484 MYR5,Myo1e,Myr3 myosin IE GO:0003779 actin binding 25576 14-3-3e1,MGC93547,Ywhah -

(Ser739) Antibody-SL10016R
SunLong Biotech Co.,LTD Tel: 0086-571- 56623320 Fax:0086-571- 56623318 E-mail:[email protected] www.sunlongbiotech.com Rabbit Anti-phospho-ZCWCC1 (Ser739) antibody SL10016R Product Name: phospho-ZCWCC1 (Ser739) Chinese Name: 磷酸化ZCWCC1抗体 AC004542.C22.1.; p-MORC2(Ser739); phospho-ZCWCC1(Ser739); CW type with coiled coil domain 1; KIAA0852; ZCW3; ZCWCC1; Zinc finger; Zinc finger CW type Alias: coiled coil domain protein 1; Zinc finger CW type with coiled coil domain 1; Zing finger CW type 3 zinc finger CW-type coiled-coil domain protein 1; MORC family CW-type zine finger 2; MORC2. Organism Species: Rabbit Clonality: Polyclonal React Species: Human,Mouse,Rat, WB=1:500-2000ELISA=1:500-1000IHC-P=1:400-800IHC-F=1:400-800ICC=1:100- 500IF=1:100-500(Paraffin sections need antigen repair) Applications: not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. Molecular weight: 114kDa Cellular localization: The nucleuscytoplasmic Form: Lyophilizedwww.sunlongbiotech.com or Liquid Concentration: 1mg/ml KLH conjugated synthesised phosphopeptide derived from human MORC2 around the immunogen: phosphorylation site of Ser739:KR(p-S)VA Lsotype: IgG Purification: affinity purified by Protein A Storage Buffer: 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year Storage: when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C. -

Plekhg5-Regulated Autophagy of Synaptic Vesicles Reveals a Pathogenic Mechanism in Motoneuron Disease
Thomas Jefferson University Jefferson Digital Commons Department of Medicine Faculty Papers Department of Medicine 12-1-2017 Plekhg5-regulated autophagy of synaptic vesicles reveals a pathogenic mechanism in motoneuron disease. Patrick Lüningschrör University Hospital Würzburg; University of Bielefeld Beyenech Binotti Max Planck Institute for Biophysical Chemistry, Göttingen Benjamin Dombert University Hospital Würzburg Follow this and additional works at: https://jdc.jefferson.edu/medfp Peter Heimann Univ Persityart of of the Bielef Neureldology Commons AngelLet usPerez-Lar knowa how access to this document benefits ouy University of Bielefeld Recommended Citation Lüningschrör, Patrick; Binotti, Beyenech; Dombert, Benjamin; Heimann, Peter; Perez-Lara, Angel; See next page for additional authors Slotta, Carsten; Thau-Habermann, Nadine; von Collenberg, Cora R.; Karl, Franziska; Damme, Markus; Horowitz, Arie; Maystadt, Isabelle; Füchtbauer, Annette; Füchtbauer, Ernst-Martin; Jablonka, Sibylle; Blum, Robert; Üçeyler, Nurcan; Petri, Susanne; Kaltschmidt, Barbara; Jahn, Reinhard; Kaltschmidt, Christian; and Sendtner, Michael, "Plekhg5-regulated autophagy of synaptic vesicles reveals a pathogenic mechanism in motoneuron disease." (2017). Department of Medicine Faculty Papers. Paper 220. https://jdc.jefferson.edu/medfp/220 This Article is brought to you for free and open access by the Jefferson Digital Commons. The Jefferson Digital Commons is a service of Thomas Jefferson University's Center for Teaching and Learning (CTL). The Commons is a showcase for Jefferson books and journals, peer-reviewed scholarly publications, unique historical collections from the University archives, and teaching tools. The Jefferson Digital Commons allows researchers and interested readers anywhere in the world to learn about and keep up to date with Jefferson scholarship. This article has been accepted for inclusion in Department of Medicine Faculty Papers by an authorized administrator of the Jefferson Digital Commons. -

Genome-Wide DNA Methylation Analysis on C-Reactive Protein Among Ghanaians Suggests Molecular Links to the Emerging Risk of Cardiovascular Diseases ✉ Felix P
www.nature.com/npjgenmed ARTICLE OPEN Genome-wide DNA methylation analysis on C-reactive protein among Ghanaians suggests molecular links to the emerging risk of cardiovascular diseases ✉ Felix P. Chilunga 1 , Peter Henneman2, Andrea Venema2, Karlijn A. C. Meeks 3, Ana Requena-Méndez4,5, Erik Beune1, Frank P. Mockenhaupt6, Liam Smeeth7, Silver Bahendeka8, Ina Danquah9, Kerstin Klipstein-Grobusch10,11, Adebowale Adeyemo 3, Marcel M.A.M Mannens2 and Charles Agyemang1 Molecular mechanisms at the intersection of inflammation and cardiovascular diseases (CVD) among Africans are still unknown. We performed an epigenome-wide association study to identify loci associated with serum C-reactive protein (marker of inflammation) among Ghanaians and further assessed whether differentially methylated positions (DMPs) were linked to CVD in previous reports, or to estimated CVD risk in the same population. We used the Illumina Infinium® HumanMethylation450 BeadChip to obtain DNAm profiles of blood samples in 589 Ghanaians from the RODAM study (without acute infections, not taking anti-inflammatory medications, CRP levels < 40 mg/L). We then used linear models to identify DMPs associated with CRP concentrations. Post-hoc, we evaluated associations of identified DMPs with elevated CVD risk estimated via ASCVD risk score. We also performed subset analyses at CRP levels ≤10 mg/L and replication analyses on candidate probes. Finally, we assessed for biological relevance of our findings in public databases. We subsequently identified 14 novel DMPs associated with CRP. In post-hoc evaluations, we found 1234567890():,; that DMPs in PC, BTG4 and PADI1 showed trends of associations with estimated CVD risk, we identified a separate DMP in MORC2 that was associated with CRP levels ≤10 mg/L, and we successfully replicated 65 (24%) of previously reported DMPs. -

Mutations Suggests Both Loss-Of-Function and Gain-Of-Function Effects Morag A
© 2021. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2021) 14, dmm047225. doi:10.1242/dmm.047225 RESEARCH ARTICLE Hearing impairment due to Mir183/96/182 mutations suggests both loss-of-function and gain-of-function effects Morag A. Lewis1,2,*, Francesca Di Domenico1, Neil J. Ingham1,2, Haydn M. Prosser2 and Karen P. Steel1,2 ABSTRACT birth. However, this is not the cause of the hearing loss; even before The microRNA miR-96 is important for hearing, as point mutations in the onset of normal hearing, homozygote hair cells fail to mature humans and mice result in dominant progressive hearing loss. Mir96 both morphologically and physiologically, remaining in their is expressed in sensory cells along with Mir182 and Mir183, but the immature state, and heterozygote hair cells show a developmental roles of these closely-linked microRNAs are as yet unknown. Here, delay. miR-96 is thus thought to be responsible for coordinating we analyse mice carrying null alleles of Mir182, and of Mir183 and hair cell maturation (Chen et al., 2014; Kuhn et al., 2011). Mir96 together to investigate their roles in hearing. We found that Overexpression of the three miRNAs also results in cochlear defects Mir183/96 heterozygous mice had normal hearing and homozygotes and hearing loss (Weston et al., 2018). The complete loss of all were completely deaf with abnormal hair cell stereocilia bundles and mature miRNAs from the inner ear results in early developmental reduced numbers of inner hair cell synapses at 4 weeks of age. defects including a severely truncated cochlear duct (Friedman Mir182 knockout mice developed normal hearing then exhibited et al., 2009; Soukup et al., 2009). -

Genetic Modifiers of Hereditary Neuromuscular Disorders
cells Article Genetic Modifiers of Hereditary Neuromuscular Disorders and Cardiomyopathy Sholeh Bazrafshan 1, Hani Kushlaf 2 , Mashhood Kakroo 1, John Quinlan 2, Richard C. Becker 1 and Sakthivel Sadayappan 1,* 1 Heart, Lung and Vascular Institute, Division of Cardiovascular Health and Disease, Department of Internal Medicine, University of Cincinnati College of Medicine, Cincinnati, OH 45267, USA; [email protected] (S.B.); [email protected] (M.K.); [email protected] (R.C.B.) 2 Department of Neurology and Rehabilitation Medicine, Neuromuscular Center, University of Cincinnati Gardner Neuroscience Institute, University of Cincinnati College of Medicine, Cincinnati, OH 45267, USA; [email protected] (H.K.); [email protected] (J.Q.) * Correspondence: [email protected]; Tel.: +1-513-558-7498 Abstract: Novel genetic variants exist in patients with hereditary neuromuscular disorders (NMD), including muscular dystrophy. These patients also develop cardiac manifestations. However, the association between these gene variants and cardiac abnormalities is understudied. To determine genetic modifiers and features of cardiac disease in NMD patients, we have reviewed electronic medical records of 651 patients referred to the Muscular Dystrophy Association Care Center at the University of Cincinnati and characterized the clinical phenotype of 14 patients correlating with their next-generation sequencing data. The data were retrieved from the electronic medical records of the 14 patients included in the current study and comprised neurologic and cardiac phenotype and genetic reports which included comparative genomic hybridization array and NGS. Novel associations were uncovered in the following eight patients diagnosed with Limb-girdle Muscular Dystrophy, Bethlem Myopathy, Necrotizing Myopathy, Charcot-Marie-Tooth Disease, Peripheral Citation: Bazrafshan, S.; Kushlaf, H.; Kakroo, M.; Quinlan, J.; Becker, R.C.; Polyneuropathy, and Valosin-containing Protein-related Myopathy. -

Biological Role and Disease Impact of Copy Number Variation in Complex Disease
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2014 Biological Role and Disease Impact of Copy Number Variation in Complex Disease Joseph Glessner University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, and the Genetics Commons Recommended Citation Glessner, Joseph, "Biological Role and Disease Impact of Copy Number Variation in Complex Disease" (2014). Publicly Accessible Penn Dissertations. 1286. https://repository.upenn.edu/edissertations/1286 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/1286 For more information, please contact [email protected]. Biological Role and Disease Impact of Copy Number Variation in Complex Disease Abstract In the human genome, DNA variants give rise to a variety of complex phenotypes. Ranging from single base mutations to copy number variations (CNVs), many of these variants are neutral in selection and disease etiology, making difficult the detection of true common orar r e frequency disease-causing mutations. However, allele frequency comparisons in cases, controls, and families may reveal disease associations. Single nucleotide polymorphism (SNP) arrays and exome sequencing are popular assays for genome-wide variant identification. oT limit bias between samples, uniform testing is crucial, including standardized platform versions and sample processing. Bases occupy single points while copy variants occupy segments. -

Selection Signatures Scan in Several Italian Sheep Breeds Identifies Genes Influencing Micronutrient Metabolism S
Selection signatures scan in several Italian sheep breeds identifies genes influencing micronutrient metabolism S. Sorbolini1, C. Dimauro1, M. Cellesi1, F. Pilla2, N.P.P. Macciotta1 and the BIOVITA Consortium. 1Università di Sassari, Dipartimento di Agraria, Viale Italia 39, 07100 Sassari, Italy. 1Università del Studi del Molise, Dipartimento Agricoltura Ambiente Alimenti, Via F. de Sanctis s.n.c. 86100 Campobasso, Italy. [email protected] (Corresponding author) Summary Animal physiological functions involve enzymes and cofactors. Many of these substances can not be synthesized by the body, but derive from the diet. An example are micronutrients, that strongly affect production performance, whose requirements are often met by the farmers by adding supplements to the diet. The aim of this study was to investigate the genetic variability of Italian sheep breeds searching for possible selective sweeps that harbor genes involved in the metabolism of micronutrients in. SNP A sample of 496 sheep belonging to 20 breeds farmed in Italy were genotyped with the Illumina Ovine 50K beadchip. Data were analysed by canonical discriminant analysis (CDA). Forty SNP located in regions of the genome containing loci involved in the metabolism of vitamins and minerals were detected. in particular, genes linked to the metabolism of vitamins and minerals such as Selenocysteine Lyase (SCLY), calcium sensing receptor (CASR), Solute Carrier Family 23 Member 1 (SLC23A1) and Thiamine Triphosphatase (THTPA) were highlighted. Keywords: selection signatures, micronutrients, sheep, Canonical Discriminant Analysis Introduction Nutritional status is of particular importance for productive performances in livestock. Growth, milk production, reproduction depend on a wide range of essential nutrients such as amino acids, fatty acids, vitamins and minerals. -

Neuropathic MORC2 Mutations Perturb GHKL Atpase Dimerization Dynamics and Epigenetic Silencing by Multiple Structural Mechanisms
Neuropathic MORC2 mutations perturb GHKL ATPase dimerization dynamics and epigenetic silencing by multiple structural mechanisms The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Douse, Christopher H., Stuart Bloor, Yangci Liu, Maria Shamin, Iva A. Tchasovnikarova, Richard T. Timms, Paul J. Lehner, and Yorgo Modis. 2018. “Neuropathic MORC2 mutations perturb GHKL ATPase dimerization dynamics and epigenetic silencing by multiple structural mechanisms.” Nature Communications 9 (1): 651. doi:10.1038/s41467-018-03045-x. http://dx.doi.org/10.1038/ s41467-018-03045-x. Published Version doi:10.1038/s41467-018-03045-x Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:35014920 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA ARTICLE DOI: 10.1038/s41467-018-03045-x OPEN Neuropathic MORC2 mutations perturb GHKL ATPase dimerization dynamics and epigenetic silencing by multiple structural mechanisms Christopher H. Douse 1, Stuart Bloor2, Yangci Liu1, Maria Shamin1, Iva A. Tchasovnikarova 2,3, Richard T. Timms2,4, Paul J. Lehner2 & Yorgo Modis 1 1234567890():,; Missense mutations in MORC2 cause neuropathies including spinal muscular atrophy and Charcot–Marie–Tooth disease. We recently identified MORC2 as an effector of epigenetic silencing by the human silencing hub (HUSH). Here we report the biochemical and cellular activities of MORC2 variants, alongside crystal structures of wild-type and neuropathic forms of a human MORC2 fragment comprising the GHKL-type ATPase module and CW-type zinc finger. -

Genetics of Distal Hereditary Motor Neuropathies
GENETICSOFDISTALHEREDITARY MOTOR NEUROPATHIES By alexander peter drew A thesis submitted for the Degree of Doctor of Philosophy Supervised by Professor Garth A. Nicholson Dr. Ian P. Blair Faculty of Medicine University of Sydney 2012 statement No part of the work described in this thesis has been submitted in fulfilment of the requirements for any other academic degree or qualification. Except where due acknowledgement has been made, all experimental work was performed by the author. Alexander Peter Drew CONTENTS acknowledgements ............................. i summary .................................... ii list of figures ................................ v list of tables ................................ viii acronyms and abbreviations ..................... xi publications ................................. xiv 1 literature review ........................... 1 1.1 Molecular genetics and mechanisms of disease in Distal Hereditary Motor Neuropathies . .1 1.1.1 Small heat shock protein family . .2 1.1.2 Dynactin 1 (DCTN1).....................9 1.1.3 Immunoglobulin mu binding protein 2 gene (IGHMBP2) 11 1.1.4 Senataxin (SETX)....................... 14 1.1.5 Glycyl-tRNA synthase (GARS)............... 16 1.1.6 Berardinelli-Seip congenital lipodystrophy 2 (SEIPIN) gene (BSCL2)......................... 18 1.1.7 ATPase, Cu2+-transporting, alpha polypeptide gene (ATP7A) 20 1.1.8 Pleckstrin homology domain-containing protein, G5 gene (PLEKHG5)........................... 21 1.1.9 Transient receptor potential cation channel, V4 gene (TRPV4) 22 1.1.10 DYNC1H1 ........................... 23 1.1.11 Clinical variability in dHMN . 24 1.1.12 Common disease mechanisms in dHMN . 29 2 general materials and methods ................. 32 2.1 General materials and reagents . 32 2.1.1 Reagents and Enzymes . 32 2.1.2 Equipment . 33 2.1.3 Plasticware . 33 2.2 Study participants . 34 2.3 DNA methods . -

Genetic and Functional Investigation of Inherited Neuropathies
GENETIC AND FUNCTIONAL INVESTIGATION OF INHERITED NEUROPATHIES Ellen Cottenie MRC Centre for Neuromuscular Diseases, UCL Institute of Neurology Supervisors: Professor Mary M. Reilly, Professor Henry Houlden and Professor Mike Hanna Thesis submitted for the degree of Doctor of Philosophy University College London 2015 1 Declaration I, Ellen Cottenie, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. 2 Abstract With the discovery of next generation sequencing techniques the landscape of pathogenic gene discovery has shifted drastically over the last ten years. For the purpose of this thesis, focus was applied on finding genetic causes of inherited neuropathies, mainly Charcot-Marie-Tooth disease, by using both old and new genetic techniques and the accompanying functional investigations to prove the pathogenicity of these variants. Mutations in ATPase 6, the first mitochondrially encoded gene responsible for an isolated neuropathy, were found in five families with CMT2 by a traditional Sanger sequencing approach. The same approach was used to expand the phenotype associated with FIG4 mutations, known as CMT4J. Compound heterozygous mutations were found in a patient with a proximal and asymmetric weakness and rapid deterioration of strength in a single limb, mimicking CIDP. Several appropriate cohorts were screened for mutations in candidate genes with the traditional Sanger sequencing approach; however, no new pathogenic genes were found. In the case of the HINT1 gene, the originally stated frequency of 11% could not be replicated and a founder effect was suggested, underlying the importance of considering the ethnic background of a patient when screening for mutations in neuropathy-related genes. -
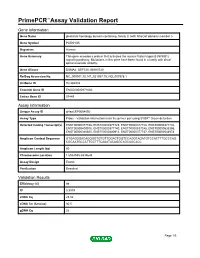
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name pleckstrin homology domain containing, family G (with RhoGef domain) member 5 Gene Symbol PLEKHG5 Organism Human Gene Summary This gene encodes a protein that activates the nuclear factor kappa B (NFKB1) signaling pathway. Multations in this gene have been found in a family with distal spinal muscular atrophy. Gene Aliases DSMA4, GEF720, KIAA0720 RefSeq Accession No. NC_000001.10, NT_021937.19, NG_007978.1 UniGene ID Hs.284232 Ensembl Gene ID ENSG00000171680 Entrez Gene ID 57449 Assay Information Unique Assay ID qHsaCEP0054092 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000377748, ENST00000377725, ENST00000377728, ENST00000377732, ENST00000400915, ENST00000377740, ENST00000537245, ENST00000535355, ENST00000340850, ENST00000400913, ENST00000377737, ENST00000544978 Amplicon Context Sequence GTGAGGGACAGGGGTGTGTTGGACTGGTCCAGGTAGATGTCCACTTTGCCCAG CGCAATGCCCTTCCTTTCAAATACAGGCAGCAGCACC Amplicon Length (bp) 60 Chromosome Location 1:6534555-6534644 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 98 R2 0.9979 cDNA Cq 25.02 cDNA Tm (Celsius) 80.5 gDNA Cq 26 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report PLEKHG5, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of