Gunning Et Al Febslett
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Interactions of Insecticidal Spider Peptide Neurotoxins with Insect Voltage- and Neurotransmitter-Gated Ion Channels
Interactions of insecticidal spider peptide neurotoxins with insect voltage- and neurotransmitter-gated ion channels (Molecular representation of - HXTX-Hv1c including key binding residues, adapted from Gunning et al, 2008) PhD Thesis Monique J. Windley UTS 2012 CERTIFICATE OF AUTHORSHIP/ORIGINALITY I certify that the work in this thesis has not previously been submitted for a degree nor has it been submitted as part of requirements for a degree except as fully acknowledged within the text. I also certify that the thesis has been written by me. Any help that I have received in my research work and the preparation of the thesis itself has been acknowledged. In addition, I certify that all information sources and literature used are indicated in the thesis. Monique J. Windley 2012 ii ACKNOWLEDGEMENTS There are many people who I would like to thank for contributions made towards the completion of this thesis. Firstly, I would like to thank my supervisor Prof. Graham Nicholson for his guidance and persistence throughout this project. I would like to acknowledge his invaluable advice, encouragement and his neverending determination to find a solution to any problem. He has been a valuable mentor and has contributed immensely to the success of this project. Next I would like to thank everyone at UTS who assisted in the advancement of this research. Firstly, I would like to acknowledge Phil Laurance for his assistance in the repair and modification of laboratory equipment. To all the laboratory and technical staff, particulary Harry Simpson and Stan Yiu for the restoration and sourcing of equipment - thankyou. I would like to thank Dr Mike Johnson for his continual assistance, advice and cheerful disposition. -

Comparative Analyses of Venoms from American and African Sicarius Spiders That Differ in Sphingomyelinase D Activity
This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright Author's personal copy Toxicon 55 (2010) 1274–1282 Contents lists available at ScienceDirect Toxicon journal homepage: www.elsevier.com/locate/toxicon Comparative analyses of venoms from American and African Sicarius spiders that differ in sphingomyelinase D activity Pamela A. Zobel-Thropp*, Melissa R. Bodner 1, Greta J. Binford Department of Biology, Lewis and Clark College, 0615 SW Palatine Hill Road, Portland, OR 97219, USA article info abstract Article history: Spider venoms are cocktails of toxic proteins and peptides, whose composition varies at Received 27 August 2009 many levels. Understanding patterns of variation in chemistry and bioactivity is funda- Received in revised form 14 January 2010 mental for understanding factors influencing variation. The venom toxin sphingomyeli- Accepted 27 January 2010 nase D (SMase D) in sicariid spider venom (Loxosceles and Sicarius) causes dermonecrotic Available online 8 February 2010 lesions in mammals. Multiple forms of venom-expressed genes with homology to SMase D are expressed in venoms of both genera. -

ANA CAROLINA MARTINS WILLE.Pdf
UNIVERSIDADE FEDERAL DO PARANÁ ANA CAROLINA MARTINS WILLE AVALIAÇÃO DA ATIVIDADE DE FOSFOLIPASE-D RECOMBINANTE DO VENENO DA ARANHA MARROM (Loxosceles intermedia) SOBRE A PROLIFERAÇÃO, INFLUXO DE CÁLCIO E METABOLISMO DE FOSFOLIPÍDIOS EM CÉLULAS TUMORAIS. CURITIBA 2014 i Wille, Ana Carolina Martins Avaliação da atividade de fosfolipase-D recombinante do veneno da aranha marrom (Loxosceles intermedia) sobre a proliferação, influxo de cálcio e metabolismo de fosfolipídios em células tumorais Curitiba, 2014. 217p. Tese (Doutorado) – Universidade Federal do Paraná – UFPR 1.veneno de aranha marrom. 2. fosfolipase-D. 3.proliferação celular. 4.metabolismo de lipídios. 5.influxo de cálcio. ANA CAROLINA MARTINS WILLE AVALIAÇÃO DA ATIVIDADE DE FOSFOLIPASE-D RECOMBINANTE DO VENENO DA ARANHA MARROM (Loxosceles intermedia) SOBRE A PROLIFERAÇÃO, INFLUXO DE CÁLCIO E METABOLISMO DE FOSFOLIPÍDIOS EM CÉLULAS TUMORAIS. Tese apresentada como requisito à obtenção do grau de Doutor em Biologia Celular e Molecular, Curso de Pós- Graduação em Biologia Celular e Molecular, Setor de Ciências Biológicas, Universidade Federal do Paraná. Orientador(a): Dra. Andrea Senff Ribeiro Co-orientador: Dr. Silvio Sanches Veiga CURITIBA 2014 ii O desenvolvimento deste trabalho foi possível devido ao apoio financeiro do Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), a Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação Araucária e SETI-PR. iii Dedico este trabalho àquela que antes da sua existência foi o grande sonho que motivou minha vida. Sonho que foi a base para que eu escolhesse uma profissão e um trabalho. À você, minha amada filha GIOVANNA, hoje minha realidade, dedico todo meu trabalho. iv Dedico também este trabalho ao meu amado marido, amigo, professor e co- orientador Dr. -

Arthropod Envenomation
Arthropod Envenomation Michael R. Loomis, DVM, MA, DACZM North Carolina Zoological Park Hymenoptera Envenomation Order Hymenoptera Family Vespidae- wasps Family Formicidae- ants Familt Mutillidae- velvet ants Family Apidae- bees • Stinger is a modified ovipositor Bee and Wasp Venom Components • Proteins, peptides and • Apitoxin – 52% Melitten (potent anti- amines inflammatory agent that – Phospholipase increases production of cortisol) – Histamine – 10-12% Phospholipase A2 – Bradykinin – 2-5% Aldolapin (blocks – cyclooxygenase) Acetylcholine – 1-3% Hyuronidase – Dopamine – 0.5-2% Histamine – Seratonin – 1-2% Dopamine and noradrenaline – Mast cell degranulating – 2% Protease-inhibitors peptide – Apamine increases cortisol – Mastoparan production, mild neurotoxin Ant Venom Components • Fire ants- 95% alkaloid (Unique among ants) • Most other ants, similar to bee and wasp venom • Harvester ant venom contains a hemolysin Venom Toxicity Family Common Name LD 50 (mg/kg) Apidae Honey bee 2.8 Mutillidae Velvet ant 71.0 Vespidae Paper wasp 2.4 Vespidae Yellowjacket 3.5 Formicidae Harvester ant 0.66 Formicidae Maricopa Harvester ant 0.12 Morbidity and Mortality Bees and Wasps • In US, 9.3 million ant • 17-56% produce local stings and 1 million reactions stings of other • Hymenoptera/year 1-2% produce generalized reactions • More deaths/year than any other type of • 5% seek medical care envenomation • 30-120 deaths from • Most deaths are the wasp and bee result of Anaphylaxis stings/year Local Reactions • Pain • Edema which may extend 10 cm from -

Structural Venomics Reveals Evolution of a Complex Venom by Duplication and Diversification of an Ancient Peptide-Encoding Gene
Structural venomics reveals evolution of a complex venom by duplication and diversification of an ancient peptide-encoding gene Sandy S. Pinedaa,b,1,2,3,4, Yanni K.-Y. China,c,1, Eivind A. B. Undheimc,d,e, Sebastian Senffa, Mehdi Moblic, Claire Daulyf, Pierre Escoubasg, Graham M. Nicholsonh, Quentin Kaasa, Shaodong Guoa, Volker Herziga,5, John S. Mattickb,6, and Glenn F. Kinga,2 aInstitute for Molecular Bioscience, The University of Queensland, St Lucia, QLD 4072, Australia; bGarvan-Weizmann Centre for Cellular Genomics, Garvan Institute of Medical Research, Darlinghurst, Sydney, NSW 2010, Australia; cCentre for Advanced Imaging, The University of Queensland, St Lucia, QLD 4072, Australia; dCentre for Biodiversity Dynamics, Department of Biology, Norwegian University of Science and Technology, 7491 Trondheim, Norway; eCentre for Ecological & Evolutionary Synthesis, Department of Biosciences, University of Oslo, 0316 Oslo, Norway; fThermo Fisher Scientific, 91941 Courtaboeuf Cedex, France; gUniversity of Nice Sophia Antipolis, 06000 Nice, France; and hSchool of Life Sciences, University of Technology Sydney, Broadway, NSW 2007, Australia Edited by Adriaan Bax, National Institutes of Health, Bethesda, MD, and approved March 18, 2020 (received for review August 21, 2019) Spiders are one of the most successful venomous animals, with N-terminal strand is sometimes present (12). The cystine knot more than 48,000 described species. Most spider venoms are comprises a “ring” formed by two disulfide bonds and the in- dominated by cysteine-rich peptides with a diverse range of phar- tervening sections of the peptide backbone, with a third disulfide macological activities. Some spider venoms contain thousands of piercing the ring to create a pseudoknot (11). -

Venom Composition and Bioactivity from the Eurasian Assassin Bug Rhynocoris Iracundus
biomedicines Article Hexapod Assassins’ Potion: Venom Composition and Bioactivity from the Eurasian Assassin Bug Rhynocoris iracundus Nicolai Rügen 1, Timothy P. Jenkins 2, Natalie Wielsch 3, Heiko Vogel 4 , Benjamin-Florian Hempel 5,6 , Roderich D. Süssmuth 5 , Stuart Ainsworth 7, Alejandro Cabezas-Cruz 8 , Andreas Vilcinskas 1,9,10 and Miray Tonk 9,10,* 1 Department of Bioresources, Fraunhofer Institute for Molecular Biology and Applied Ecology, Ohlebergsweg 12, 35392 Giessen, Germany; [email protected] (N.R.); [email protected] (A.V.) 2 Department of Biotechnology and Biomedicine, Technical University of Denmark, 2800 Kongens Lyngby, Denmark; [email protected] 3 Research Group Mass Spectrometry/Proteomics, Max Planck Institute for Chemical Ecology, Hans-Knoell-Strasse 8, 07745 Jena, Germany; [email protected] 4 Department of Entomology, Max Planck Institute for Chemical Ecology, Hans-Knöll-Straße 8, 07745 Jena, Germany; [email protected] 5 Department of Chemistry, Technische Universität Berlin, Strasse des 17. Juni 124, 10623 Berlin, Germany; [email protected] (B.-F.H.); [email protected] (R.D.S.) 6 BIH Center for Regenerative Therapies BCRT, Charité—Universitätsmedizin Berlin, 13353 Berlin, Germany 7 Centre for Snakebite Research and Interventions, Department of Tropical Disease Biology, Liverpool School of Tropical Medicine, Liverpool L3 5QA, UK; [email protected] 8 Citation: Rügen, N.; Jenkins, T.P.; UMR BIPAR, Laboratoire de Santé Animale, Anses, INRAE, Ecole Nationale Vétérinaire d’Alfort, Wielsch, N.; Vogel, H.; Hempel, B.-F.; F-94700 Maisons-Alfort, France; [email protected] 9 Institute for Insect Biotechnology, Justus Liebig University of Giessen, Heinrich-Buff-Ring 26-32, Süssmuth, R.D.; Ainsworth, S.; 35392 Giessen, Germany Cabezas-Cruz, A.; Vilcinskas, A.; 10 LOEWE Centre for Translational Biodiversity Genomics (LOEWE-TBG), Senckenberganlage 25, Tonk, M. -

Pilbara Project Short-Range Endemic Invertebrate Fauna Survey
Short-Range Endemic Invertebrate Fauna Report: FerrAus Pilbara Project Prepared for FerrAus Ltd Final Report 5HY October 2010 Phoenix Environmental Sciences Pty Ltd 1 Short-range Endemic Invertebrate Fauna Survey Final Report FerrAus Pilbara Project FerrAus Ltd Short-range Endemic Invertebrate Fauna Survey 3URMHFW)HUU$XV3LOEDUD3URMHFW )LQDO5HSRUW5HY October 2010 Authors: Conor O’Neill and Jarrad Clark Reviewer: Melanie White Prepared for FerrAus Ltd Prepared by: Phoenix Environmental Sciences Pty Ltd © 2010 Phoenix Environmental Sciences Pty Ltd The information contained in this report is solely for the use of the Client for the purpose in which it has been prepared and Phoenix Environmental Sciences Pty Ltd accepts no responsibility for use beyond this purpose. Any person or organisation wishing to quote or reproduce any section of this report may only do so with the written permission of Phoenix Environmental Sciences Pty Ltd or FerrAus Ltd. Phoenix Environmental Sciences Pty Ltd 1/511 Wanneroo Road BALCATTA WA 6021 P: 08 9345 1608 F: 08 6313 0680 E: [email protected] Project code: 952-DC-FER-SRE Phoenix Environmental Sciences Pty Ltd i Short-range Endemic Invertebrate Fauna Survey Final Report FerrAus Pilbara Project FerrAus Ltd TABLE OF CONTENTS EXECUTIVE SUMMARY ................................................................................................................................... iv 1.0 INTRODUCTION ....................................................................................................................................... -

FUDMA Journal of Sciences (FJS) Vol. 4 No. 2, June, 2020, Pp 92 - 100 92 EFFECT of PHYSICO-CHEMICAL… Akpan, Et Al., FJS
FUDMA Journal of Sciences (FJS) EFFECT OF PHYSICO-CHEMICAL… ISSNAkpan, online: et al., 2616 -1370 FJS ISSN print: 2645 - 2944 Vol. 4 No. 2, June, 2020, pp 92 -100 DOI: https://doi.org/10.33003/fjs -2020-0402-206 EFFECT OF PHYSICO-CHEMICAL PARAMETERS ON THE ABUNDANCE AND DIVERSITY OF TERMITES AND OTHER ARTHROPODS IN TERMITE MOUNDS IN UYO, AKWA IBOM STATE, NIGERIA. *1Akpan, Akaninyene Udoh, 2Ojianwuna, Chioma Cynthia, 1Ubulom, Peace Mayen Edwin, 1Clement Ameh Yaro, 1Oboho, Diligent Efiong. 1Entomology Unit – Department of Animal and Environmental Biology, University of Uyo, Uyo, Akwa Ibom State, Nigeria. 2Department of Animal and Environmental Biology, Delta State University, Abraka. Delta State. Nigeria *Corresponding author e-mail: [email protected] ABSTRACT Termites are generally regarded as pests, although they have some beneficial roles to play in the ecosystem, particularly in the soil. This study was conducted between January 2018 and April 2018, to determine the effect of physico-chemical parametrs on abundance and diversity of termites and other arthropods in termite mounds in Uinversity of Uyo Community. Soil samples were randomly collected from six termite mounds from two sites for physiochemical parameters analysis and these were temperature, pH, moisture content, nitrogen, phosphorus, magnesium, copper, sodium, potassium, manganese and iron.. The termites and other arthropods were preserved in 70% ethanol. Temperature and moisture content, copper, sodium and iron were significant. The results revealed that the physicochemical parameters affected the termite species abundance as station 1 (539) had relatively more of the termite species than station 2 (551), and also affected the diversity of the termites as station 1 (0.89) had relatively more diversity of the termites than station 2 (0.66). -
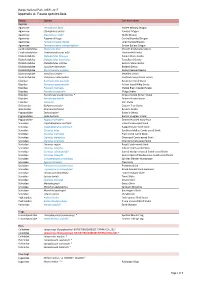
Report-Mungo National Park-Appendix A
Mungo National Park, NSW, 2017 Appendix A: Fauna species lists Family Species Common name Reptiles Agamidae Ctenophorus fordi Mallee Military Dragon Agamidae Ctenophorus pictus Painted Dragon Agamidae Diporiphora nobbi Nobbi Dragon Agamidae Pogona vitticeps Central Bearded Dragon Agamidae Tympanocryptis lineata Lined Earless Dragon Agamidae Tympanocryptis tetraporophora Eyrean Earless Dragon Carphodactylidae Nephrurus levis Smooth Knob-tailed Gecko Carphodactylidae Underwoodisaurus milii Thick-tailed Gecko Diplodactylidae Diplodactylus furcosus Ranges Stone Gecko Diplodactylidae Diplodactylus tessellatus Tessellated Gecko Diplodactylidae Diplodactylus vittatus Eastern Stone Gecko Diplodactylidae Lucasium damaeum Beaded Gecko Diplodactylidae Rhynchoedura ormsbyi Eastern Beaked Gecko Diplodactylidae Strophurus elderi ~ Jewelled Gecko Diplodactylidae Strophurus intermedius Southern Spiny-tailed Gecko Elapidae Brachyurophis australis Australian Coral Snake Elapidae Demansia psammophis Yellow-faced Whip Snake Elapidae Parasuta nigriceps Mallee Black-headed Snake Elapidae Pseudechis australis Mulga Snake Elapidae Pseudonaja aspidorhyncha * Strap-snouted Brown Snake Elapidae Pseudonaja textilis Eastern Brown Snake Elapidae Suta suta Curl Snake Gekkonidae Gehyra versicolor Eastern Tree Gecko Gekkonidae Heteronotia binoei Bynoe's Gecko Pygopodidae Delma butleri Butler's Delma Pygopodidae Lialis burtonis Burton's Legless Lizard Pygopodidae Pygopus schraderi Eastern Hooded Scaly-foot Scincidae Cryptoblepharus australis Inland Snake-eyed Skink Scincidae -

Diversification of a Single Ancestral Gene Into a Successful Toxin Superfamily in Highly Venomous Australian Funnel-Web Spiders
Pineda et al. BMC Genomics 2014, 15:177 http://www.biomedcentral.com/1471-2164/15/177 RESEARCH ARTICLE Open Access Diversification of a single ancestral gene into a successful toxin superfamily in highly venomous Australian funnel-web spiders Sandy S Pineda1†, Brianna L Sollod2,7†, David Wilson1,3,8†, Aaron Darling1,9, Kartik Sunagar4,5, Eivind A B Undheim1,6, Laurence Kely6, Agostinho Antunes4,5, Bryan G Fry1,6* and Glenn F King1* Abstract Background: Spiders have evolved pharmacologically complex venoms that serve to rapidly subdue prey and deter predators. The major toxic factors in most spider venoms are small, disulfide-rich peptides. While there is abundant evidence that snake venoms evolved by recruitment of genes encoding normal body proteins followed by extensive gene duplication accompanied by explosive structural and functional diversification, the evolutionary trajectory of spider-venom peptides is less clear. Results: Here we present evidence of a spider-toxin superfamily encoding a high degree of sequence and functional diversity that has evolved via accelerated duplication and diversification of a single ancestral gene. The peptides within this toxin superfamily are translated as prepropeptides that are posttranslationally processed to yield the mature toxin. The N-terminal signal sequence, as well as the protease recognition site at the junction of the propeptide and mature toxin are conserved, whereas the remainder of the propeptide and mature toxin sequences are variable. All toxin transcripts within this superfamily exhibit a striking cysteine codon bias. We show that different pharmacological classes of toxins within this peptide superfamily evolved under different evolutionary selection pressures. Conclusions: Overall, this study reinforces the hypothesis that spiders use a combinatorial peptide library strategy to evolve a complex cocktail of peptide toxins that target neuronal receptors and ion channels in prey and predators. -

Araneae: Mygalomorphae: Actinopodidae: Missulena) from the Pilbara Region, Western Australia
Zootaxa 3637 (5): 521–540 ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ Article ZOOTAXA Copyright © 2013 Magnolia Press ISSN 1175-5334 (online edition) http://dx.doi.org/10.11646/zootaxa.3637.5.2 http://zoobank.org/urn:lsid:zoobank.org:pub:447D8DF5-F922-4B3A-AC43-A85225E56C57 New species of Mouse Spiders (Araneae: Mygalomorphae: Actinopodidae: Missulena) from the Pilbara region, Western Australia DANILO HARMS1, 2, 3, 4 & VOLKER W. FRAMENAU1, 2, 3 1 School of Animal Biology, The University of Western Australia, 35 Stirling Highway, Crawley, Western Australia 6009, Australia. 2 Department of Terrestrial Zoology, Western Australian Museum, Locked Bag 49, Welshpool DC, Western Australia 6986, Australia. 3 Phoenix Environmental Sciences Pty Ltd, 1/511 Wanneroo Road, Balcatta, Western Australia 6021, Australia. 4 Corresponding author. E-mail: [email protected] Abstract Two new species of Mouse Spiders, genus Missulena, from the Pilbara region in Western Australia are described based on morphological features of males. Missulena faulderi sp. nov. and Missulena langlandsi sp. nov. are currently known from a small area in the southern Pilbara only. Mitochondrial cytochrome c oxidase subunit I (COI) sequence divergence failed in clearly delimiting species in Missulena, but provided a useful, independent line of evidence for taxonomic work in addition to morphology. Key words: taxonomy, systematics, barcoding, mitochondrial DNA, short-range endemism, Actinopus, Plesiolena Introduction The Actinopodidae Simon, 1892 is a small family of mygalomorph spiders with a Gondwanan distribution that includes three genera: Actinopus Perty, 1833 (27 species), Missulena Walckenaer, 1805 (11 species) and Plesiolena Goloboff & Platnick, 1987 (two species). Actinopus and Plesiolena are known only from South and Central America (Platnick 2012). -

Five Papers on Fossil and Extant Spiders
BEITR. ARANEOL., 13 (2020) Joerg Wunderlich FIVE PAPERS ON FOSSIL AND EXTANT SPIDERS BEITR. ARANEOL., 13 (2020: 1–176) FIVE PAPERS ON FOSSIL AND EXTANT SPIDERS NEW AND RARE FOSSIL SPIDERS (ARANEAE) IN BALTIC AND BUR- MESE AMBERS AS WELL AS EXTANT AND SUBRECENT SPIDERS FROM THE WESTERN PALAEARCTIC AND MADAGASCAR, WITH NOTES ON SPIDER PHYLOGENY, EVOLUTION AND CLASSIFICA- TION JOERG WUNDERLICH, D-69493 Hirschberg, e-mail: [email protected]. Website: www.joergwunderlich.de. – Here a digital version of this book can be found. © Publishing House, author and editor: Joerg Wunderlich, 69493 Hirschberg, Germany. BEITRAEGE ZUR ARANEOLOGIE (BEITR. ARANEOL.), 13. ISBN 978-3-931473-19-8 The papers of this volume are available on my website. Print: Baier Digitaldruck GmbH, Heidelberg. 1 BEITR. ARANEOL., 13 (2020) Photo on the book cover: Dorsal-lateral aspect of the male tetrablemmid spider Elec- troblemma pinnae n. sp. in Burmit, body length 1.5 mm. See the photo no. 17 p. 160. Fossil spider of the year 2020. Acknowledgements: For corrections of parts of the present manuscripts I thank very much my dear wife Ruthild Schöneich. For the professional preparation of the layout I am grateful to Angelika and Walter Steffan in Heidelberg. CONTENTS. Papers by J. WUNDERLICH, with the exception of the paper p. 22 page Introduction and personal note………………………………………………………… 3 Description of four new and few rare spider species from the Western Palaearctic (Araneae: Dysderidae, Linyphiidae and Theridiidae) …………………. 4 Resurrection of the extant spider family Sinopimoidae LI & WUNDERLICH 2008 (Araneae: Araneoidea) ……………………………………………………………...… 19 Note on fossil Atypidae (Araneae) in Eocene European ambers ………………… 21 New and already described fossil spiders (Araneae) of 20 families in Mid Cretaceous Burmese amber with notes on spider phylogeny, evolution and classification; by J.