George De Hevesy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Nobel Laureate George De Hevesy (1885-1966) - Universal Genius and Father of Nuclear Medicine Niese S* Am Silberblick 9, 01723 Wilsdruff, Germany
Open Access SAJ Biotechnology LETTER ISSN: 2375-6713 The Nobel Laureate George de Hevesy (1885-1966) - Universal Genius and Father of Nuclear Medicine Niese S* Am Silberblick 9, 01723 Wilsdruff, Germany *Corresponding author: Niese S, Am Silberblick 9, 01723 Wilsdruff, Germany, Tel: +49 35209 22849, E-mail: [email protected] Citation: Niese S, The Nobel Laureate George de Hevesy (1885-1966) - Universal Genius and Father of Nuclear Medicine. SAJ Biotechnol 5: 102 Article history: Received: 20 March 2018, Accepted: 29 March 2018, Published: 03 April 2018 Abstract The scientific work of the universal genius the Nobel Laureate George de Hevesy who has discovered and developed news in physics, chemistry, geology, biology and medicine is described. Special attention is given to his work in life science which he had done in the second half of his scientific career and was the base of the development of nuclear medicine. Keywords: George de Hevesy; Radionuclides; Nuclear Medicine Introduction George de Hevesy has founded Radioanalytical Chemistry and Nuclear Medicine, discovered the element hafnium and first separated stable isotopes. He was an inventor in many disciplines and his interest was not only focused on the development and refinement of methods, but also on the structure of matter and its changes: atoms, molecules, cells, organs, plants, animals, men and cosmic objects. He was working under complicated political situation in Europe in the 20th century. During his stay in Germany, Austria, Hungary, Switzerland, Denmark, and Sweden he wrote a lot papers in German. In 1962 he edited a large part of his articles in a collection where German papers are translated in English [1]. -
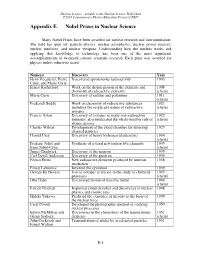
Appendix E Nobel Prizes in Nuclear Science
Nuclear Science—A Guide to the Nuclear Science Wall Chart ©2018 Contemporary Physics Education Project (CPEP) Appendix E Nobel Prizes in Nuclear Science Many Nobel Prizes have been awarded for nuclear research and instrumentation. The field has spun off: particle physics, nuclear astrophysics, nuclear power reactors, nuclear medicine, and nuclear weapons. Understanding how the nucleus works and applying that knowledge to technology has been one of the most significant accomplishments of twentieth century scientific research. Each prize was awarded for physics unless otherwise noted. Name(s) Discovery Year Henri Becquerel, Pierre Discovered spontaneous radioactivity 1903 Curie, and Marie Curie Ernest Rutherford Work on the disintegration of the elements and 1908 chemistry of radioactive elements (chem) Marie Curie Discovery of radium and polonium 1911 (chem) Frederick Soddy Work on chemistry of radioactive substances 1921 including the origin and nature of radioactive (chem) isotopes Francis Aston Discovery of isotopes in many non-radioactive 1922 elements, also enunciated the whole-number rule of (chem) atomic masses Charles Wilson Development of the cloud chamber for detecting 1927 charged particles Harold Urey Discovery of heavy hydrogen (deuterium) 1934 (chem) Frederic Joliot and Synthesis of several new radioactive elements 1935 Irene Joliot-Curie (chem) James Chadwick Discovery of the neutron 1935 Carl David Anderson Discovery of the positron 1936 Enrico Fermi New radioactive elements produced by neutron 1938 irradiation Ernest Lawrence -

Facts and Figures 2013
Facts and Figures 201 3 Contents The University 2 World ranking 4 Academic pedigree 6 Areas of impact 8 Research power 10 Spin-outs 12 Income 14 Students 16 Graduate careers 18 Alumni 20 Faculties and Schools 22 Staff 24 Estates investment 26 Visitor attractions 28 Widening participation 30 At a glance 32 1 The University of Manchester Our Strategic Vision 2020 states our mission: “By 2020, The University of Manchester will be one of the top 25 research universities in the world, where all students enjoy a rewarding educational and wider experience; known worldwide as a place where the highest academic values and educational innovation are cherished; where research prospers and makes a real difference; and where the fruits of scholarship resonate throughout society.” Our core goals 1 World-class research 2 Outstanding learning and student experience 3 Social responsibility 2 3 World ranking The quality of our teaching and the impact of our research are the cornerstones of our success. 5 The Shanghai Jiao Tong University UK Academic Ranking of World ranking Universities assesses the best teaching and research universities, and in 2012 we were ranked 40th in the world. 7 World European UK European Year Ranking Ranking Ranking ranking 2012 40 7 5 2010 44 9 5 2005 53 12 6 2004* 78* 24* 9* 40 Source: 2012 Shanghai Jiao Tong University World Academic Ranking of World Universities ranking *2004 ranking refers to the Victoria University of Manchester prior to the merger with UMIST. 4 5 Academic pedigree Nobel laureates 1900 JJ Thomson , Physics (1906) We attract the highest calibre researchers and Ernest Rutherford , Chemistry (1908) teachers, boasting 25 Nobel Prize winners among 1910 William Lawrence Bragg , Physics (1915) current and former staff and students. -

Guide to the James Franck Papers 1882-1966
University of Chicago Library Guide to the James Franck Papers 1882-1966 © 2006 University of Chicago Library Table of Contents Acknowledgments 3 Descriptive Summary 3 Information on Use 3 Access 3 Citation 3 Biographical Note 4 Scope Note 15 Related Resources 21 Subject Headings 21 INVENTORY 22 Series I: Correspondence 22 Series II: Manuscripts 51 Subseries 1: Physics - work in Germany and Denmark, 1905-1934 51 Subseries 2: Physics - work in United States, 1935-1958 53 Subseries 3: Biophysics - work on Photosynthesis at Johns Hopkins, 1935-193855 Subseries 4: Biophysics - work on Photosynthesis at the University of Chicago,55 1938-48 Subseries 5: Biophysics - work on Photosynthesis after 1948 55 Subseries 6: General Articles and Talks on Science 71 Subseries 7: Papers by other scientists 72 Subseries 8: Notes, memoranda and fragments 76 Subseries 9: Atomic Scientists' Movement, 1944-1953 76 Subseries 10: Franck Memorial Symposium, May 12-13, 1966 79 Series III: Tape Recordings and Photographs 80 Subseries 1: Tape recordings 80 Subseries 2: Hertha Sponer's photograph album, Göttingen, 1920-1933 80 Series IV: Personal Documents and Memorabilia 90 Subseries 1: Documents 90 Subseries 2: Clippings 93 Subseries 3: Biographies and Obituaries 94 Subseries 4: Memorabilia; Scrolls, Certificates, Medals, Mementos 96 Series V: Robert Platzman's Editorial Papers for the "Selected Works of James98 Franck" Series VI: Addenda 103 Subseries 1: Correspondence between James Franck and his nephew and Dr. Heinz104 Kallman Subseries 2: Oversize 105 Descriptive Summary Identifier ICU.SPCL.FRANCK Title Franck, James. Papers Date 1882-1966 Size 20.5 linear feet (29 boxes) Repository Special Collections Research Center University of Chicago Library 1100 East 57th Street Chicago, Illinois 60637 U.S.A. -

CHEMICAL HERITAGE FOUNDATION GEORGE A. OLAH Transcript
CHEMICAL HERITAGE FOUNDATION GEORGE A. OLAH Transcript of an Interview Conducted by James G. Traynham and Arnold Thackray at Loker Hydrocarbon Research Institute Los Angeles, California on 3 February 2000 (With Subsequent Corrections and Additions) This interview has been designated as Free Access. One may view, quote from, cite, or reproduce the oral history with the permission of CHF. Please note: Users citing this interview for purposes of publication are obliged under the terms of the Chemical Heritage Foundation Oral History Program to credit CHF using the format below: George A. Olah, interview by James G. Traynham and Arnold Thackray at Loker Hydrocarbon Research Institute, Los Angeles, California, 3 February 2000 (Philadelphia: Chemical Heritage Foundation, Oral History Transcript # 0190). Chemical Heritage Foundation Oral History Program 315 Chestnut Street Philadelphia, Pennsylvania 19106 GEORGE A. OLAH 1927 Born in Budapest, Hungary, on 22 May Education 1945 B.S., organic chemistry, Technical University, Budapest 1949 Ph.D., organic chemistry, Technical University, Budapest Professional Experience Technical University, Budapest, Hungary 1949-1954 Assistant Professor to Associate Professor of Organic Chemistry Hungarian Academy of Sciences 1954-1956 Head of Department of Organic Chemistry and Associate Scientific Director of Central Research Institute The Dow Chemical Company 1957-1964 Senior Research Scientist Case Western Reserve University 1965-1967 Professor and Chairman, Department of Chemistry 1967-1969 Chairman of Combined Departments of Chemistry (Case Institute and Western Reserve University) 1967-1977 C. F. Mabery Distinguished Professor of Research in Chemistry University of Southern California 1977-1977 Professor of Chemistry and Scientific Director, Hydrocarbon Research Institute 1980-present Distinguished Professor of Chemistry 1983-present Donald P. -

Nobel Prize Awards in Radiochemistry
Radiochim. Acta 100, 509–521 (2012) / DOI 10.1524/ract.2012.1953 © by Oldenbourg Wissenschaftsverlag, München Nobel Prize awards in Radiochemistry By J.-P. Adloff∗ University of Strasbourg, 63 Rue Saint Urbain, 67100 Strasbourg, France Dedicated to the memory of late Karl H. Lieser, Gerhard L. Stöcklin and Alfred P. Wolf with whom the author shared the editorial work of Radiochimica Acta from 1977 to 1995 (Received October 10, 2011; accepted in revised form January 19, 2012) (Published online March 26, 2012) Nobel Prize / Chemistry / Physics Summary. In 1996 the Editors of Radiochimica Acta brought out a special volume of the journal to celebrate the hundredth anniversary of the discovery of radioactivity [1]. On the occasion of the 50th anniversary of Radiochimica Acta, which follows closely upon the centenary of Marie Curie’s second Nobel Prize in 1911, the author has the privilege to informally review “Radiochemistry and Nobel Prize Awards”, including discoveries of radioelements and new fields in chemistry based on radiochemical methods. 1. The beginning The Nobel Prizes in Physics and Chemistry were estab- lished in 1901, six years after the discovery of radioactivity and three years after the discoveries of the elements polo- Fig. 1. Antoine Henri Becquerel (1852–1908). nium and radium. They are awarded by Kungliga Veten- skapakademien (the Royal Swedish Academy of Sciences) on the basis of proposals made by respective Committees rays when he thought the subject was exhausted. By the end on Physics and Chemistry, which receive recommendations of 1897 radioactivity was something of a dead horse: it was from Swedish and foreign scientists [2]. -
List of Nobel Laureates 1
List of Nobel laureates 1 List of Nobel laureates The Nobel Prizes (Swedish: Nobelpriset, Norwegian: Nobelprisen) are awarded annually by the Royal Swedish Academy of Sciences, the Swedish Academy, the Karolinska Institute, and the Norwegian Nobel Committee to individuals and organizations who make outstanding contributions in the fields of chemistry, physics, literature, peace, and physiology or medicine.[1] They were established by the 1895 will of Alfred Nobel, which dictates that the awards should be administered by the Nobel Foundation. Another prize, the Nobel Memorial Prize in Economic Sciences, was established in 1968 by the Sveriges Riksbank, the central bank of Sweden, for contributors to the field of economics.[2] Each prize is awarded by a separate committee; the Royal Swedish Academy of Sciences awards the Prizes in Physics, Chemistry, and Economics, the Karolinska Institute awards the Prize in Physiology or Medicine, and the Norwegian Nobel Committee awards the Prize in Peace.[3] Each recipient receives a medal, a diploma and a monetary award that has varied throughout the years.[2] In 1901, the recipients of the first Nobel Prizes were given 150,782 SEK, which is equal to 7,731,004 SEK in December 2007. In 2008, the winners were awarded a prize amount of 10,000,000 SEK.[4] The awards are presented in Stockholm in an annual ceremony on December 10, the anniversary of Nobel's death.[5] As of 2011, 826 individuals and 20 organizations have been awarded a Nobel Prize, including 69 winners of the Nobel Memorial Prize in Economic Sciences.[6] Four Nobel laureates were not permitted by their governments to accept the Nobel Prize. -

Image-Brochure-LNLM-2020-LQ.Pdf
NOBEL LAUREATES PARTICIPATING IN LINDAU EVENTS SINCE 1951 Peter Agre | George A. Akerlof | Kurt Alder | Zhores I. Alferov | Hannes Alfvén | Sidney Altman | Hiroshi Amano | Philip W. Anderson | Christian B. Anfinsen | Edward V. Appleton | Werner Arber | Frances H. Arnold | Robert J. Aumann | Julius Axelrod | Abhijit Banerjee | John Bardeen | Barry C. Barish | Françoise Barré-Sinoussi | Derek H. R. Barton | Nicolay G. Basov | George W. Beadle | J. Georg Bednorz | Georg von Békésy |Eric Betzig | Bruce A. Beutler | Gerd Binnig | J. Michael Bishop | James W. Black | Elizabeth H. Blackburn | Patrick M. S. Blackett | Günter Blobel | Konrad Bloch | Felix Bloch | Nicolaas Bloembergen | Baruch S. Blumberg | Niels Bohr | Max Born | Paul Boyer | William Lawrence Bragg | Willy Brandt | Walter H. Brattain | Bertram N. Brockhouse | Herbert C. Brown | James M. Buchanan Jr. | Frank Burnet | Adolf F. Butenandt | Melvin Calvin Thomas R. Cech | Martin Chalfie | Subrahmanyan Chandrasekhar | Pavel A. Cherenkov | Steven Chu | Aaron Ciechanover | Albert Claude | John Cockcroft | Claude Cohen- Tannoudji | Leon N. Cooper | Carl Cori | Allan M. Cormack | John Cornforth André F. Cournand | Francis Crick | James Cronin | Paul J. Crutzen | Robert F. Curl Jr. | Henrik Dam | Jean Dausset | Angus S. Deaton | Gérard Debreu | Petrus Debye | Hans G. Dehmelt | Johann Deisenhofer Peter A. Diamond | Paul A. M. Dirac | Peter C. Doherty | Gerhard Domagk | Esther Duflo | Renato Dulbecco | Christian de Duve John Eccles | Gerald M. Edelman | Manfred Eigen | Gertrude B. Elion | Robert F. Engle III | François Englert | Richard R. Ernst Gerhard Ertl | Leo Esaki | Ulf von Euler | Hans von Euler- Chelpin | Martin J. Evans | John B. Fenn | Bernard L. Feringa Albert Fert | Ernst O. Fischer | Edmond H. Fischer | Val Fitch | Paul J. -

24 August 2013 Seminar Held
PROCEEDINGS OF THE NOBEL PRIZE SEMINAR 2012 (NPS 2012) 0 Organized by School of Chemistry Editor: Dr. Nabakrushna Behera Lecturer, School of Chemistry, S.U. (E-mail: [email protected]) 24 August 2013 Seminar Held Sambalpur University Jyoti Vihar-768 019 Odisha Organizing Secretary: Dr. N. K. Behera, School of Chemistry, S.U., Jyoti Vihar, 768 019, Odisha. Dr. S. C. Jamir Governor, Odisha Raj Bhawan Bhubaneswar-751 008 August 13, 2013 EMSSSEM I am glad to know that the School of Chemistry, Sambalpur University, like previous years is organizing a Seminar on "Nobel Prize" on August 24, 2013. The Nobel Prize instituted on the lines of its mentor and founder Alfred Nobel's last will to establish a series of prizes for those who confer the “greatest benefit on mankind’ is widely regarded as the most coveted international award given in recognition to excellent work done in the fields of Physics, Chemistry, Physiology or Medicine, Literature, and Peace. The Prize since its introduction in 1901 has a very impressive list of winners and each of them has their own story of success. It is heartening that a seminar is being organized annually focusing on the Nobel Prize winning work of the Nobel laureates of that particular year. The initiative is indeed laudable as it will help teachers as well as students a lot in knowing more about the works of illustrious recipients and drawing inspiration to excel and work for the betterment of mankind. I am sure the proceeding to be brought out on the occasion will be highly enlightening. -

Radioactivity
Radioactivity Lecture 3 The Discovery of Radioactivity Science and Applications First Indications of new Phenomena • Henri Becquerel discovered 1896 that Uranium blackens a photographic film or silver-photo emulsion. He concluded that there must be a kind of unknown light • Marie and Pierre Curie looked for other materials that may emit this new kind of light. They found Pitchblende. They discovered a time dependence of the emitted radiation and a change of nature of radiation that they saw as indication for new elements, Radium and Polonium, which they extracted by chemical methods. • They developed new experimental methods to detect radiation, they coined the term Radioactivity and introduced a unit for the intensity “Curie”, which represents the number of emitted radiation by 1 g of Radium. It corresponds to 3.71010 events/s, but says nothing about the origin or nature of the radiation. Observations • Radioactivity produces heat (decay heat) • Radioactivity can emanate as gas (Radon) • Radioactivity darkens chemical emulsions • Radioactivity generates fluorescence • Radioactivity generates thermoluminescence • Radioactivity causes chemical reactions • Radioactivity causes burns and other physiological changes • Radioactivity can kill (bacteria, mice) • Radioactivity is temperature independent Discovery Conclusions Curies expectation and explanation of chemical conversion Four times higher activity than Uran, suspicion was there must be something else, a different element? Pitchblende Uraninite UO2 The Curie analyzed and converted several tons of Pitchblende a highly radioactive material that was a left-over from uranium mining. A tedious chemical effort to extract Radium and Polonium as decay products from the Uranium in the material. During that process they were exposed to enormous amounts of radiation, still observed on their notes, working and living quarters, even in the kitchen. -

CHEMISTRY International July-September 2019 Volume 41 No
CHEMISTRY International The News Magazine of IUPAC July-September 2019 Volume 41 No. 3 Special IUPAC 100 INTERNATIONAL UNION OFBrought to you by | IUPACA Glance The International at Union The of Pure Union and Applied History Chemistry PURE AND APPLIED CHEMISTRY Authenticated Download Date | 7/9/19 4:46 PM Special IUPAC100 e embarked on the adventure of researching IUPAC’s his- International CHEMISTRY tory a few years ago, inspired by the looming IUPAC 100 The News Magazine of the anniversary in 2019 and the desire of the French National International Union of Pure and W Committee to host that year’s Congress and General Assembly in Applied Chemistry (IUPAC) Paris. The proposal to host the fiftieth IUPAC General Assembly and All information regarding notes for contributors, sub- forty-seventh Congress in Paris was received and approved by IUPAC scriptions, Open Access, back volumes and orders is Council during its assembly in 2013. Soon after, Jean-Pierre Vairon, a available online at www.degruyter.com/ci member of the organizing committee of the IUPAC 2019 Congress, contacted Danielle Fauque, and together we started to think about Managing Editor: special symposia devoted to the history of IUPAC. At the Congress in Fabienne Meyers 2015 in Busan, Korea, the idea was met with enthusiasm and interest IUPAC, c/o Department of Chemistry from Natalia Tarasova, then president of the Union, and the project Boston University of this special issue was formed with Fabienne Meyers, Chemistry Metcalf Center for Science and Engineering International editor. We also met with Christopher Brett later that 590 Commonwealth Ave. -

Nobel Prizes List from 1901
Nature and Science, 4(3), 2006, Ma, Nobel Prizes Nobel Prizes from 1901 Ma Hongbao East Lansing, Michigan, USA, Email: [email protected] The Nobel Prizes were set up by the final will of Alfred Nobel, a Swedish chemist, industrialist, and the inventor of dynamite on November 27, 1895 at the Swedish-Norwegian Club in Paris, which are awarding to people and organizations who have done outstanding research, invented groundbreaking techniques or equipment, or made outstanding contributions to society. The Nobel Prizes are generally awarded annually in the categories as following: 1. Chemistry, decided by the Royal Swedish Academy of Sciences 2. Economics, decided by the Royal Swedish Academy of Sciences 3. Literature, decided by the Swedish Academy 4. Peace, decided by the Norwegian Nobel Committee, appointed by the Norwegian Parliament, Stortinget 5. Physics, decided by the Royal Swedish Academy of Sciences 6. Physiology or Medicine, decided by Karolinska Institutet Nobel Prizes are widely regarded as the highest prize in the world today. As of November 2005, a total of 776 Nobel Prizes have been awarded, 758 to individuals and 18 to organizations. [Nature and Science. 2006;4(3):86- 94]. I. List of All Nobel Prize Winners (1901 – 2005): 31. Physics, Philipp Lenard 32. 1906 - Chemistry, Henri Moissan 1. 1901 - Chemistry, Jacobus H. van 't Hoff 33. Literature, Giosuè Carducci 2. Literature, Sully Prudhomme 34. Medicine, Camillo Golgi 3. Medicine, Emil von Behring 35. Medicine, Santiago Ramón y Cajal 4. Peace, Henry Dunant 36. Peace, Theodore Roosevelt 5. Peace, Frédéric Passy 37. Physics, J.J. Thomson 6. Physics, Wilhelm Conrad Röntgen 38.