Clinical Consequences of Axonal Injury in Traumatic Brain Injury
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Slow Axonal Transport and Presynaptic Targeting of Clathrin Packets
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.20.958140; this version posted February 20, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Slow Axonal Transport and Presynaptic Targeting of Clathrin Packets Archan Ganguly 1,2, Florian Wernert3, Sébastien Phan 2,4, Daniela Boassa 2,4, Utpal Das 1,2, Rohan Sharma 1,2, Ghislaine Caillol 3, Xuemei Han 5, John R. Yates III 5, Mark H. Ellisman 2,4, Christophe Leterrier 3, and Subhojit Roy 1,2 * 1 Department of Pathology, University of California, San Diego, La Jolla, CA 2 Department of Neurosciences, University of California, San Diego, La Jolla, CA 3 Aix Marseille Université, CNRS, INP UMR7051, NeuroCyto, Marseille, France 4 National Center for Microscopy and Imaging Research, University of California, San Diego, La Jolla, CA 5 Department of Cell Biology, The Scripps Research Institute, La Jolla, CA * Correspondence: [email protected] SUMMARY Clathrin has established roles in endocytosis, with clathrin-cages enclosing membrane infoldings, followed by rapid disassembly and reuse of clathrin-monomers. In neurons, clathrin synthesized in cell- bodies is conveyed via slow axonal transport – enriching at presynapses – however, mechanisms underlying transport/targeting are unknown, and its role at synapses is debated. Combining live- imaging, mass-spectrometry (MS), Apex-labeled EM-tomography and super-resolution, we found that unlike dendrites where clathrin assembles and disassembles as expected, axons contain stable ‘clathrin transport-packets’ that move intermittently with an anterograde bias, conveying endocytosis-related proteins. -

Axonal Transport and the Eye
Br J Ophthalmol: first published as 10.1136/bjo.60.8.547 on 1 August 1976. Downloaded from Brit. 7. Ophthal. (1976) 60, 547 Editorial: Axonal transport and the eye Since the earliest days of histology, it has been and proteins, and are predominantly found in the known that the neurons have a large amount of particulate fraction of nerve homogenates. The basophilic material in the cell body. More recently slower transported material is mainly soluble this has been shown to be due to masses of rough protein (McEwen and Grafstein, I968; Kidwai and endoplasmic reticulum which are responsible for a Ochs, I969; Sj6strand, 1970). Much of the former high rate of protein synthesis of neurons. Since the and more than 6o per cent of the latter is probably earliest days of the microscopy of living cells in neurotubular protein (tubulin), and the neurotubules vitro, particulate movement within these cells has are thought to play a major part in axonal transport been recognized, particularly in neurons (Nakai, (Schmitt and Samson, I968). The neurotubules I964). The last 30 years have seen the gradual have a similar structure to actin, and are linked to accumulation of the knowledge required to explain high-energy phosphate compounds. Myosin-like these two observations. In the last io years in proteins are also found in axoplasm. It is suggested particular, the development of radioactive tracer that moving material might in part roll along the techniques has revealed much information on the neurotubules, or that the neurotubules themselves movement of protein and other substances within might move by a mechanism involving myosin- axons, a process termed axonal transport (Barondes, guanosine triphosphate-neurotubule cross bonds I967). -

Vesicular Glycolysis Provides On-Board Energy for Fast Axonal Transport
Vesicular Glycolysis Provides On-Board Energy for Fast Axonal Transport Diana Zala,1,2,3 Maria-Victoria Hinckelmann,1,2,3 Hua Yu,1,2,3 Marcel Menezes Lyra da Cunha,1,4 Ge´ raldine Liot,1,2,3,6 Fabrice P. Cordelie` res,1,5 Sergio Marco,1,4 and Fre´ de´ ric Saudou1,2,3,* 1Institut Curie, 91405 Orsay, France 2CNRS UMR3306, 91405 Orsay, France 3INSERM U1005, 91405 Orsay, France 4INSERM U759, 91405 Orsay, France 5IBiSA Cell and Tissue Imaging Facility, Institut Curie, 91405 Orsay, France 6Present address: Institut Curie, CNRS UMR 3347, INSERM U1021, Universite´ Paris-Sud 11, 91405 Orsay, France *Correspondence: [email protected] http://dx.doi.org/10.1016/j.cell.2012.12.029 SUMMARY and Vale, 2009), nerve cells must have energy sources distrib- uted throughout their axons for these molecular motors to Fast axonal transport (FAT) requires consistent function. energy over long distances to fuel the molecular Glucose is the major source of brain energy and is required for motors that transport vesicles. We demonstrate normal brain function. Glucose breakdown occurs in the cytosol that glycolysis provides ATP for the FAT of vesicles. through the glycolytic pathway that converts glucose into pyru- Although inhibiting ATP production from mitochon- vate and generates free energy (Ames, 2000). The net energetic dria did not affect vesicles motility, pharmacological balance of glycolysis is relatively low, with two ATP being produced from one molecule of glucose. In contrast to glycol- or genetic inhibition of the glycolytic enzyme GAPDH ysis, mitochondria produce 34 ATP molecules (Ames, 2000) reduced transport in cultured neurons and in and provide most of the energy used by the brain. -
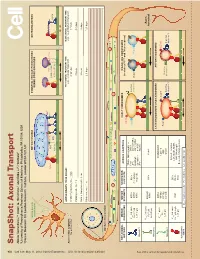
Snapshot: Axonal Transport Alison Twelvetrees,1,2 Adam G
950 Cell SnapShot: Axonal Transport 149 Alison Twelvetrees,1,2 Adam G. Hendricks,1 and Erika L.F. Holzbaur1 , May11, 2012©2012Elsevier Inc. DOI 10.1016/j.cell.2012.05.001 1University of Pennsylvania Perelman School of Medicine, Philadelphia, PA 19104, USA 2Cancer Research UK London Research Institute, London WC2A 3LY, UK Mixed polarity MITOCHONDRIA SYNAPTIC VESICLE PRECURSORS NEUROFILAMENTS microtubules DENSE CORE GRANULES Syntabulin Miro FEZ1 Rab3 Liprin-α Dynein TRAKs JIP1 KBP DENN/MADD KIF5 KIF5A KIF1Bα KIF1A/KIF1Bβ MICROTUBULE AXONS DRAWN TO 5X SCALE: FAST AXONAL TRANSPORT TIME SLOW AXONAL TRANSPORT TIME Axon initial segment e.g., vesicular transport e.g., neurolament transport Selective lter Inhibitory interneuron - 1 mm 4 minutes 3 hours Purkinje cell - 36 mm 2 hours 4.5 days Retinal Ganglion cell - 5 cm 3 hours 6 days Motor neuron - 1 m 2.5 days 125 days Axonal cross-section + Uniform polarity microtubules + + KEY AXONAL MOTOR MOTOR NONMOTOR AXONAL ADAPTORS EARLY ENDOSOMES SIGNALING ENDOSOMES MOTORS PROPERTIES SUBUNITS SUBUNITS (contain p75NTR and Trk receptors) EEA1 TRAKs (Milton) and Miro V 0.8 µm/s KIF5A KLCs Fez1 DISC1 Inactive max JIPs Slp1/CRMP-2 Rab5 KIF5B (not always kinesin-2 Rab7 Rab5 Fs 5-7 pN Huntingtin LIS1/NUDEL See online version for legend and references. Lr 1-2 µm KIF5C required) Syntabulin mNUDC Kinesin-1 APP HSc70 V 0.43 µm/s Axon max KIF3A/B KAP3 Fodrin Fs 5 pN KIF3C terminal Lr 0.45 µm Kinesin-2 LATE ENDOSOMES AND LYSOSOMES AUTOPHAGOSOMES KIF1A DENN/MADD V 1 µm/s max KIF1Bα Liprin-α LAMP1 LC3-II Fs - KIF1Bβ KBP Rab7 Lr 1 µm Inactive Kinesin-3 KIF13B PIP3BP Inactive kinesin-2 Inactive kinesin-1 kinesin-1 Dynactin complex V 0.8 µm/s DICs max LIS1, NudE, NuDEL Fs 1 pN, 6 pN DHC DLICs HAP1/Huntingtin Lr 1 µm DLCs Bicaudal-D family proteins Cytoplasmic dynein Vmax= Maximal velocity Fs = Stall force Lr = Length of run SnapShot: Axonal Transport Alison Twelvetrees,1,2 Adam G. -

A Student's Guide to Neural Circuit Tracing
fnins-13-00897 August 23, 2019 Time: 18:23 # 1 REVIEW published: 27 August 2019 doi: 10.3389/fnins.2019.00897 A Student’s Guide to Neural Circuit Tracing Christine Saleeba1,2†, Bowen Dempsey3†, Sheng Le1, Ann Goodchild1 and Simon McMullan1* 1 Neurobiology of Vital Systems Node, Faculty of Medicine and Health Sciences, Macquarie University, Sydney, NSW, Australia, 2 The School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, United Kingdom, 3 CNRS, Hindbrain Integrative Neurobiology Laboratory, Neuroscience Paris-Saclay Institute (Neuro-PSI), Université Paris-Saclay, Gif-sur-Yvette, France The mammalian nervous system is comprised of a seemingly infinitely complex network of specialized synaptic connections that coordinate the flow of information through it. The field of connectomics seeks to map the structure that underlies brain function at resolutions that range from the ultrastructural, which examines the organization of individual synapses that impinge upon a neuron, to the macroscopic, which examines gross connectivity between large brain regions. At the mesoscopic level, distant and local connections between neuronal populations are identified, Edited by: providing insights into circuit-level architecture. Although neural tract tracing techniques Vaughan G. Macefield, have been available to experimental neuroscientists for many decades, considerable Baker Heart and Diabetes Institute, methodological advances have been made in the last 20 years due to synergies between Australia the fields of molecular biology, virology, microscopy, computer science and genetics. As Reviewed by: Patrice G. Guyenet, a consequence, investigators now enjoy an unprecedented toolbox of reagents that University of Virginia, United States can be directed against selected subpopulations of neurons to identify their efferent and Eberhard Weihe, University of Marburg, Germany afferent connectomes. -

Anterograde Neuronal Circuit Tracers Derived from Herpes Simplex Virus 1: Development, Application, and Perspectives
International Journal of Molecular Sciences Review Anterograde Neuronal Circuit Tracers Derived from Herpes Simplex Virus 1: Development, Application, and Perspectives 1,2 1,2 1,2 3 1,2, 4,5, , Dong Li , Hong Yang , Feng Xiong , Xiangmin Xu , Wen-Bo Zeng y, Fei Zhao * y and 1,2, , Min-Hua Luo * y 1 State Key Laboratory of Virology, CAS Center for Excellence in Brain Science and Intelligence Technology, Center for Biosafety Mega-Science, Wuhan Institute of Virology, Chinese Academy of Sciences, Wuhan 430071, China; [email protected] (D.L.); [email protected] (H.Y.); [email protected] (F.X.); [email protected] (W.-B.Z.) 2 University of Chinese Academy of Sciences, Beijing 100049, China 3 Department of Anatomy and Neurobiology, School of Medicine, University of California, Irvine, CA 92697-1275, USA; [email protected] 4 School of Basic Medical Sciences, Capital Medical University, Beijing 100069, China 5 Chinese Institute for Brain Research, Beijing 102206, China * Correspondence: [email protected] (F.Z.); [email protected] (M.-H.L.) These authors contribute equally. y Received: 20 July 2020; Accepted: 17 August 2020; Published: 18 August 2020 Abstract: Herpes simplex virus type 1 (HSV-1) has great potential to be applied as a viral tool for gene delivery or oncolysis. The broad infection tropism of HSV-1 makes it a suitable tool for targeting many different cell types, and its 150 kb double-stranded DNA genome provides great capacity for exogenous genes. Moreover, the features of neuron infection and neuron-to-neuron spread also offer special value to neuroscience. -

CHENEU-D-11-00020R1 Title
Elsevier Editorial System(tm) for Journal of Chemical Neuroanatomy Manuscript Draft Manuscript Number: CHENEU-D-11-00020R1 Title: A half century of experimental neuroanatomical tracing Article Type: Review Article Keywords: Tract-tracing; Fluoro-Gold; Cholera toxin; Biotinylated dextran amine; Phaseolus vulgaris- leucoagglutinin Corresponding Author: Dr. Jose Luis Lanciego, MD, PhD Corresponding Author's Institution: Center for Applied Medical Research First Author: Jose Luis Lanciego, MD, PhD Order of Authors: Jose Luis Lanciego, MD, PhD; Floris G Wouterlood, PhD Abstract: Most of our current understanding of brain function and dysfunction has its firm base in what is so elegantly called the 'anatomical substrate', i.e. the anatomical, histological, and histochemical domains within the large knowledge envelope called 'neuroscience' that further includes physiological, pharmacological, neurochemical, behavioral, genetical and clinical domains. This review focuses mainly on the anatomical domain in neuroscience. To a large degree neuroanatomical tract-tracing methods have paved the way in this domain. Over the past few decades, a great number of neuroanatomical tracers have been added to the technical arsenal to fulfill almost any experimental demand. Despite this sophisticated arsenal, the decision which tracer is best suited for a given tracing experiment still represents a difficult choice. Although this review is obviously not intended to provide the last word in the tract-tracing field, we provide a survey of the available tracing methods including some of their roots. We further summarize our experience with neuroanatomical tracers, in an attempt to provide the novice user with some advice to help this person to select the most appropriate criteria to choose a tracer that best applies to a given experimental design. -

Role of Actin Filaments in the Axonal Transport of Microtubules
The Journal of Neuroscience, December 15, 2004 • 24(50):11291–11301 • 11291 Development/Plasticity/Repair Role of Actin Filaments in the Axonal Transport of Microtubules Thomas P. Hasaka,* Kenneth A. Myers,* and Peter W. Baas Department of Neurobiology and Anatomy, Drexel University College of Medicine, Philadelphia, Pennsylvania 19129 Microtubules originate at the centrosome of the neuron and are then released for transport down the axon, in which they can move both anterogradely and retrogradely during axonal growth. It has been hypothesized that these movements occur by force generation against the actin cytoskeleton. To test this, we analyzed the movement, distribution, and orientation of microtubules in neurons pharmacolog- ically depleted of actin filaments. Actin depletion reduced but did not eliminate the anterograde movements and had no effect on the frequencyofretrogrademovements.Consistentwiththeideathatmicrotubulesmightalsomoveagainstneighboringmicrotubules,actin depletion completely inhibited the outward transport of microtubules under experimental conditions of low microtubule density. Inter- estingly, visualization of microtubule assembly shows that actin depletion actually enhances the tendency of microtubules to align with one another. Such microtubule–microtubule interactions are sufficient to orient microtubules in their characteristic polarity pattern in axons grown overnight in the absence of actin filaments. In fact, microtubule behaviors were only chaotic after actin depletion in peripheral regions of the neuron in which microtubules are normally sparse and hence lack neighboring microtubules with which they could interact. On the basis of these results, we conclude that microtubules are transported against either actin filaments or neighboring microtubules in the anterograde direction but only against other microtubules in the retrograde direction. Moreover, the transport of microtubules against one another provides a surprisingly effective option for the deployment and orientation of microtubules in the absence of actin filaments. -

Differential Loss of Bidirectional Axonal Transport with Structural Persistence Within the Same Optic Projection of the DBA/2J Glaucomatous Mouse
Differential Loss of Bidirectional Axonal Transport with Structural Persistence Within The Same Optic Projection of the DBA/2J Glaucomatous Mouse A Thesis Presented in partial fulfillment of the requirements for the degree of Masters of Science in the College of Graduate Studies of Northeast Ohio Medical University Matthew A. Smith B.A. Integrated Pharmaceutical Medicine Northeast Ohio Medical University 2014 Thesis Committee: Dr. Samuel D. Crish (advisor) Dr. Christine Crish Dr. Denise Inman Copyright Matthew A. Smith 2014 Abstract Glaucoma, the second leading cause of blindness worldwide, involves the degeneration of retinal ganglion cell bodies and their axons resulting in progressive vision loss. Much like other neurodegenerations, deficits in axonal transport are an early manifestation in the pathological progression of glaucoma. Previous studies suggest that anterograde and retrograde transport are differentially challenged and pre-degenerative in glaucoma, yet, both forms of transport have never been assessed within the same animal. We used a modified surgical procedure to assess retrograde transport while preserving the structure of the superior colliculus (SC) for both anterograde transport and immunohistochemical analysis in the same optic projection. Our findings demonstrate a 3-fold greater reduction in anterograde transport compared to retrograde transport in 9-10 month old animals. Retrograde transport remained largely intact until 13 months of age, where a reduction similar to anterograde transport was observed. Additionally, immunohistochemical staining revealed that retinal ganglion cell (RGC) axons remained intact until 13 months despite these early transport deficits. Together these data support that the RGC axonal projection remains at least semi-functional and structurally intact after anterograde transport loss in glaucoma. -
The Lifecycle of the Neuronal Microtubule Transport Machinery
This is a repository copy of The lifecycle of the neuronal microtubule transport machinery. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/158173/ Version: Accepted Version Article: Twelvetrees, A. orcid.org/0000-0002-1796-1508 (2020) The lifecycle of the neuronal microtubule transport machinery. Seminars in Cell and Developmental Biology. ISSN 1084- 9521 https://doi.org/10.1016/j.semcdb.2020.02.008 Article available under the terms of the CC-BY-NC-ND licence (https://creativecommons.org/licenses/by-nc-nd/4.0/). Reuse This article is distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivs (CC BY-NC-ND) licence. This licence only allows you to download this work and share it with others as long as you credit the authors, but you can’t change the article in any way or use it commercially. More information and the full terms of the licence here: https://creativecommons.org/licenses/ Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request. [email protected] https://eprints.whiterose.ac.uk/ The lifecycle of the neuronal microtubule transport machinery. Alison E. Twelvetrees1 1. Sheffield Institute for Translational Neuroscience, University of Sheffield, 385 Glossop Road, Sheffield S10 2HQ, UK Abstract Neurons are incredibly reliant on their cytoskeletal transport machinery. During development the cytoskeleton is the primary driver of growth and remodelling. In mature neurons the cytoskeleton keeps all components in a constant state of movement, allowing both supply of newly synthesized proteins to distal locations as well as the removal of aging proteins and organelles for recycling or degradation. -

Tracers in Neuroscience: Causation, Constraints, and Connectivity
Tracers in neuroscience: Causation, constraints, and connectivity Lauren N. Ross This paper examines tracer techniques in neuroscience, which are used to identify neural connections in the brain and nervous system. These connections capture a type of \structural connectivity" that is expected to inform our understanding of the functional nature of these tissues (Sporns 2007). This is due to the fact that neural connectivity constrains the flow of signal propagation, which is a type of causal process in neurons. This work explores how tracers are used to identify causal information, what standards they are expected to meet, the forms of causal information they provide, and how an analysis of these techniques contributes to the philosophical literature, in particular, the literature on mark transmission and mechanistic accounts of causation. 1 Introduction. In efforts to better understand the functioning of the human brain, many projects in neuroscience examine the causal process of signal propagation along neurons. In studying this causal process, a large amount of research investigates anatomical neural connections, which constrain and track the flow of these signals (Sporns 2007). These anatomical connections capture a form of \structural connectivity" that is thought to provide us with information about function, as structure informs function.1 In fact, this is a significant motivation behind the human connectome project, which aims to map all neurons and neuronal connections in the human brain (Lichtman and Sanes 2008; Sporns 2012). In some sense, this project is similar to how we might study an electronic device. If we are interested in how this device works we might start by identifying the circuit along which the electricity flows. -

Slow Axonal Transport☆ a Brown, Ohio State University, Columbus, OH, USA
Slow Axonal Transport☆ A Brown, Ohio State University, Columbus, OH, USA ã 2014 Elsevier Inc. All rights reserved. Overview 1 Methods for Studying Slow Axonal Transport 1 Accumulation Techniques 1 Pulse-Labeling Techniques 3 Imaging Techniques 3 A Brief History of Experimental Studies on Slow Axonal Transport 4 Two Distinct Subcomponents of Slow Axonal Transport 5 The Kinetics of Slow Axonal Transport 7 The Mechanism of Slow Axonal Transport 8 A Unified Perspective for Fast and Slow Axonal Transport 8 Overview Axons can be 1 m or more in length in large animals, yet they are dependent on the cell body for the synthesis of most of their components. Materials destined for the axon are transported anterogradely, toward the axon tip, and materials destined to return are transported retrogradely, toward the cell body. This bidirectional transport process, known as axonal transport, is not fundamentally different from the pathways of macromolecular and membrane traffic found in other parts of the neuron, or indeed in any eukaryotic cell, but it is unique because of the distances traveled. Kinetic studies on axonal transport in vivo using radioisotopic pulse-labeling have defined two distinct patterns of movement, termed fast and slow. Fast axonal transport represents the movement of membranous organelles, including Golgi-derived vesicles, endocytic organelles, mitochondria, peroxisomes, lysosomes, and autophagosomes. For the most part, these cargoes move rapidly and frequently at rates of up to several hundred millimeters per day, which corresponds to several micrometers per second. Some organelles move anterogradely, some retrogradely, and some (such as mitochondria) are capable of frequent reversals and move in both directions.