Differential Loss of Bidirectional Axonal Transport with Structural Persistence Within the Same Optic Projection of the DBA/2J Glaucomatous Mouse
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Raav-Based Retrograde Trans-Synaptic Labeling, Expression and Perturbation
bioRxiv preprint doi: https://doi.org/10.1101/537928; this version posted February 1, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. rAAV-based retrograde trans-synaptic tracing retroLEAP: rAAV-based retrograde trans-synaptic labeling, expression and perturbation Janine K Reinert1,#,* & Ivo Sonntag1,#,*, Hannah Sonntag2, Rolf Sprengel2, Patric Pelzer1, 1 1 1 Sascha Lessle , Michaela Kaiser , Thomas Kuner 1 Department Functional Neuroanatomy, Institute for Anatomy and Cell Biology, Heidelberg University, Heidelberg, Germany 2 Max Planck Research Group at the Institute for Anatomy and Cell Biology, at Heidelberg University, Heidelberg, Germany #These authors contributed equally to this work * Correspondence: Janine K Reinert: [email protected] Ivo Sonntag: [email protected] 1 out of 11 bioRxiv preprint doi: https://doi.org/10.1101/537928; this version posted February 1, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. rAAV-based retrograde trans-synaptic tracing Abstract Identifying and manipulating synaptically connected neurons across brain regions remains a core challenge in understanding complex nervous systems. RetroLEAP is a novel approach for retrograde trans-synaptic Labelling, Expression And Perturbation. GFP-dependent recombinase (Flp-DOG) detects trans-synaptic transfer of GFP-tetanus toxin heavy chain fusion protein (GFP-TTC) and activates expression of any gene of interest in synaptically connected cells. RetroLEAP overcomes existing limitations, is non-toxic, highly flexible, efficient, sensitive and easy to implement. Keywords: trans-synaptic, tracing, TTC, Cre-dependent, GFP-dependent flippase. -

Slow Axonal Transport and Presynaptic Targeting of Clathrin Packets
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.20.958140; this version posted February 20, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Slow Axonal Transport and Presynaptic Targeting of Clathrin Packets Archan Ganguly 1,2, Florian Wernert3, Sébastien Phan 2,4, Daniela Boassa 2,4, Utpal Das 1,2, Rohan Sharma 1,2, Ghislaine Caillol 3, Xuemei Han 5, John R. Yates III 5, Mark H. Ellisman 2,4, Christophe Leterrier 3, and Subhojit Roy 1,2 * 1 Department of Pathology, University of California, San Diego, La Jolla, CA 2 Department of Neurosciences, University of California, San Diego, La Jolla, CA 3 Aix Marseille Université, CNRS, INP UMR7051, NeuroCyto, Marseille, France 4 National Center for Microscopy and Imaging Research, University of California, San Diego, La Jolla, CA 5 Department of Cell Biology, The Scripps Research Institute, La Jolla, CA * Correspondence: [email protected] SUMMARY Clathrin has established roles in endocytosis, with clathrin-cages enclosing membrane infoldings, followed by rapid disassembly and reuse of clathrin-monomers. In neurons, clathrin synthesized in cell- bodies is conveyed via slow axonal transport – enriching at presynapses – however, mechanisms underlying transport/targeting are unknown, and its role at synapses is debated. Combining live- imaging, mass-spectrometry (MS), Apex-labeled EM-tomography and super-resolution, we found that unlike dendrites where clathrin assembles and disassembles as expected, axons contain stable ‘clathrin transport-packets’ that move intermittently with an anterograde bias, conveying endocytosis-related proteins. -

Cortical Organization: Neuroanatomical Approaches
Changes in characteristics Cortical organization: seen through phylogeny neuroanatomical Increase in the absolute and particularly approaches relative mass of the brain compared to body size Comparatively larger increase in telencephalic Study of neuronal circuitry structures particularly the cerebral cortex and microstructure Helen Barbas Expansion in number of radial units [email protected] http://www.bu.edu/neural/ Neuroinformatics Aug., 2009 From: Rakic 1 Evolution of the Nervous System Neoteny: prolongation of fetal growth How does a larger or reorganized brain come about? rate into postnatal times results in increase in the size of the human brain Environmental factors Internal factors – change in posture freedom of the hands movement of eyes from a lateral to central position EMERGENCE OF BINOCULAR VISION Crossing at the optic chiasm is complete in animals with small degree of binocular vision FREEDOM OF THE HANDS Direct projection to the pyramidal tract in animals with fine use of the hands 2 Distinct modes of radial migration: How did these changes come about in evolution? Inside-out: characterizes pattern in mammalian cortex By what mechanism? Outside-in: characterizes reptilian cortex Consequences of differential migration: differences in cortical thickness, synaptic interactions Factors contributing to the inside-out pattern reelin (glycoprotein) Addition of new cells to the mice deficient in reelin have inverted primate CNS cortical lamination cdk5 (cyclin-dependent protein kinase) mice lacking cdk5 or its activator p35 Example -

Axonal Transport and the Eye
Br J Ophthalmol: first published as 10.1136/bjo.60.8.547 on 1 August 1976. Downloaded from Brit. 7. Ophthal. (1976) 60, 547 Editorial: Axonal transport and the eye Since the earliest days of histology, it has been and proteins, and are predominantly found in the known that the neurons have a large amount of particulate fraction of nerve homogenates. The basophilic material in the cell body. More recently slower transported material is mainly soluble this has been shown to be due to masses of rough protein (McEwen and Grafstein, I968; Kidwai and endoplasmic reticulum which are responsible for a Ochs, I969; Sj6strand, 1970). Much of the former high rate of protein synthesis of neurons. Since the and more than 6o per cent of the latter is probably earliest days of the microscopy of living cells in neurotubular protein (tubulin), and the neurotubules vitro, particulate movement within these cells has are thought to play a major part in axonal transport been recognized, particularly in neurons (Nakai, (Schmitt and Samson, I968). The neurotubules I964). The last 30 years have seen the gradual have a similar structure to actin, and are linked to accumulation of the knowledge required to explain high-energy phosphate compounds. Myosin-like these two observations. In the last io years in proteins are also found in axoplasm. It is suggested particular, the development of radioactive tracer that moving material might in part roll along the techniques has revealed much information on the neurotubules, or that the neurotubules themselves movement of protein and other substances within might move by a mechanism involving myosin- axons, a process termed axonal transport (Barondes, guanosine triphosphate-neurotubule cross bonds I967). -

High-Resolution Data-Driven Model of the Mouse Connectome
RESEARCH High-resolution data-driven model of the mouse connectome Joseph E. Knox1,2, Kameron Decker Harris 2,3, Nile Graddis1, Jennifer D. Whitesell 1, Hongkui Zeng 1, Julie A. Harris1, Eric Shea-Brown 1,2, and Stefan Mihalas 1,2 1Allen Institute for Brain Science, Seattle, Washington, USA 2Applied Mathematics, University of Washington, Seattle, Washington, USA 3Computer Science and Engineering, University of Washington, Seattle, Washington, USA Keywords: Connectome, Whole-brain, Mouse an open access journal ABSTRACT Knowledge of mesoscopic brain connectivity is important for understanding inter- and intraregion information processing. Models of structural connectivity are typically constructed and analyzed with the assumption that regions are homogeneous. We instead use the Allen Mouse Brain Connectivity Atlas to construct a model of whole-brain connectivity at the scale of 100 µm voxels. The data consist of 428 anterograde tracing experiments in wild type C57BL/6J mice, mapping fluorescently labeled neuronal projections brain-wide. Inferring spatial connectivity with this dataset is underdetermined, since the approximately 2 × 105 source voxels outnumber the number of experiments. Citation: Knox, J. E., Harris, K. D., To address this issue, we assume that connection patterns and strengths vary smoothly Graddis, N., Whitesell, J. D., Zeng, H., Harris, J. A., Shea-Brown, E., & across major brain divisions. We model the connectivity at each voxel as a radial basis Mihalas, S. (2019). High-resolution data-driven model of the mouse kernel-weighted average of the projection patterns of nearby injections. The voxel model connectome. Network Neuroscience, outperforms a previous regional model in predicting held-out experiments and compared 3(1), 217–236. -

Hypothesis-Driven Structural Connectivity Analysis Supports Network Over Hierarchical Model of Brain Architecture
Hypothesis-driven structural connectivity analysis supports network over hierarchical model of brain architecture Richard H. Thompson and Larry W. Swanson1 Department of Biological Sciences, University of Southern California, Los Angeles, CA 90089 Contributed by Larry W. Swanson, June 30, 2010 (sent for review May 14, 2010) The brain is usually described as hierarchically organized, although (9) and a massive input from the SUBv (10). Only two major an alternative network model has been proposed. To help distin- brainstem inputs were identified: the dorsal raphé (DR), the guish between these two fundamentally different structure-function presumptive source of ACBdmt serotonin (11), and the inter- hypotheses, we developed an experimental circuit-tracing strategy fascicular nucleus (IF), the presumptive source of ACBdmt do- that can be applied to any starting point in the nervous system and pamine (12) that is medial to the expected ventral tegmental area then systematically expanded, and applied it to a previously obscure (VTA) itself (13). Thus, the ACBdmt receives direct inputs from dorsomedial corner of the nucleus accumbens identified functionally three massively interconnected cerebral regions implicated in as a “hedonic hot spot.” A highly topographically organized set of stress and depression, as well as restricted brainstem sites pro- connections involving expected and unexpected gray matter regions viding dopaminergic and serotonergic terminals. was identified that prominently features regions associated with Only one ACBdmt axonal output was labeled with PHAL (Fig. appetite, stress, and clinical depression. These connections are 1A; the filled purple area is a representative injection site): a arranged as a longitudinal series of circuits (closed loops). Thus, the descending pathway through the medial forebrain bundle with two results do not support a rigidly hierarchical model of nervous system clear terminal fields. -

Vesicular Glycolysis Provides On-Board Energy for Fast Axonal Transport
Vesicular Glycolysis Provides On-Board Energy for Fast Axonal Transport Diana Zala,1,2,3 Maria-Victoria Hinckelmann,1,2,3 Hua Yu,1,2,3 Marcel Menezes Lyra da Cunha,1,4 Ge´ raldine Liot,1,2,3,6 Fabrice P. Cordelie` res,1,5 Sergio Marco,1,4 and Fre´ de´ ric Saudou1,2,3,* 1Institut Curie, 91405 Orsay, France 2CNRS UMR3306, 91405 Orsay, France 3INSERM U1005, 91405 Orsay, France 4INSERM U759, 91405 Orsay, France 5IBiSA Cell and Tissue Imaging Facility, Institut Curie, 91405 Orsay, France 6Present address: Institut Curie, CNRS UMR 3347, INSERM U1021, Universite´ Paris-Sud 11, 91405 Orsay, France *Correspondence: [email protected] http://dx.doi.org/10.1016/j.cell.2012.12.029 SUMMARY and Vale, 2009), nerve cells must have energy sources distrib- uted throughout their axons for these molecular motors to Fast axonal transport (FAT) requires consistent function. energy over long distances to fuel the molecular Glucose is the major source of brain energy and is required for motors that transport vesicles. We demonstrate normal brain function. Glucose breakdown occurs in the cytosol that glycolysis provides ATP for the FAT of vesicles. through the glycolytic pathway that converts glucose into pyru- Although inhibiting ATP production from mitochon- vate and generates free energy (Ames, 2000). The net energetic dria did not affect vesicles motility, pharmacological balance of glycolysis is relatively low, with two ATP being produced from one molecule of glucose. In contrast to glycol- or genetic inhibition of the glycolytic enzyme GAPDH ysis, mitochondria produce 34 ATP molecules (Ames, 2000) reduced transport in cultured neurons and in and provide most of the energy used by the brain. -

Connectional Architecture of a Mouse Hypothalamic Circuit Node Controlling Social Behavior
Connectional architecture of a mouse hypothalamic circuit node controlling social behavior Liching Loa,b,c,1, Shenqin Yaod,1, Dong-Wook Kima,c, Ali Cetind, Julie Harrisd, Hongkui Zengd, David J. Andersona,b,c,2, and Brandon Weissbourda,b,c,2 aDivision of Biology and Biological Engineering, California Institute of Technology, Pasadena, CA 91125; bHoward Hughes Medical Institute, California Institute of Technology, Pasadena, CA 91125; cTianqiao and Chrissy Chen Institute for Neuroscience, California Institute of Technology, Pasadena, CA 91125; and dAllen Institute for Brain Science, Seattle, WA 98109 Contributed by David J. Anderson, December 12, 2018 (sent for review October 11, 2018; reviewed by Clifford B. Saper and Richard B. Simerly) Type 1 estrogen receptor-expressing neurons in the ventrolateral Examples of collateralization to multiple targets, as seen in mono- subdivision of the ventromedial hypothalamus (VMHvlEsr1) play a amine neuromodulatory systems (16, 30), are few due to limitations causal role in the control of social behaviors, including aggression. in the number of tracers that can be used simultaneously (31). Here we use six different viral-genetic tracing methods to system- Type 1 estrogen receptor (Esr1)-expressing neurons in the atically map the connectional architecture of VMHvlEsr1 neurons. ventromedial hypothalamic nucleus (VMH) control social and These data reveal a high level of input convergence and output defensive behaviors, as well as metabolic states (6, 7, 9, 32; divergence (“fan-in/fan-out”) from and to over 30 distinct brain reviewed in refs. 33–38). Unlike the ARCAgrp and the MPOAGal regions, with a high degree (∼90%) of bidirectionality, including populations, which are primarily GABAergic (24), VMHvlEsr1 both direct as well as indirect feedback. -
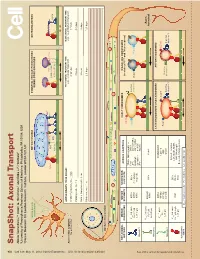
Snapshot: Axonal Transport Alison Twelvetrees,1,2 Adam G
950 Cell SnapShot: Axonal Transport 149 Alison Twelvetrees,1,2 Adam G. Hendricks,1 and Erika L.F. Holzbaur1 , May11, 2012©2012Elsevier Inc. DOI 10.1016/j.cell.2012.05.001 1University of Pennsylvania Perelman School of Medicine, Philadelphia, PA 19104, USA 2Cancer Research UK London Research Institute, London WC2A 3LY, UK Mixed polarity MITOCHONDRIA SYNAPTIC VESICLE PRECURSORS NEUROFILAMENTS microtubules DENSE CORE GRANULES Syntabulin Miro FEZ1 Rab3 Liprin-α Dynein TRAKs JIP1 KBP DENN/MADD KIF5 KIF5A KIF1Bα KIF1A/KIF1Bβ MICROTUBULE AXONS DRAWN TO 5X SCALE: FAST AXONAL TRANSPORT TIME SLOW AXONAL TRANSPORT TIME Axon initial segment e.g., vesicular transport e.g., neurolament transport Selective lter Inhibitory interneuron - 1 mm 4 minutes 3 hours Purkinje cell - 36 mm 2 hours 4.5 days Retinal Ganglion cell - 5 cm 3 hours 6 days Motor neuron - 1 m 2.5 days 125 days Axonal cross-section + Uniform polarity microtubules + + KEY AXONAL MOTOR MOTOR NONMOTOR AXONAL ADAPTORS EARLY ENDOSOMES SIGNALING ENDOSOMES MOTORS PROPERTIES SUBUNITS SUBUNITS (contain p75NTR and Trk receptors) EEA1 TRAKs (Milton) and Miro V 0.8 µm/s KIF5A KLCs Fez1 DISC1 Inactive max JIPs Slp1/CRMP-2 Rab5 KIF5B (not always kinesin-2 Rab7 Rab5 Fs 5-7 pN Huntingtin LIS1/NUDEL See online version for legend and references. Lr 1-2 µm KIF5C required) Syntabulin mNUDC Kinesin-1 APP HSc70 V 0.43 µm/s Axon max KIF3A/B KAP3 Fodrin Fs 5 pN KIF3C terminal Lr 0.45 µm Kinesin-2 LATE ENDOSOMES AND LYSOSOMES AUTOPHAGOSOMES KIF1A DENN/MADD V 1 µm/s max KIF1Bα Liprin-α LAMP1 LC3-II Fs - KIF1Bβ KBP Rab7 Lr 1 µm Inactive Kinesin-3 KIF13B PIP3BP Inactive kinesin-2 Inactive kinesin-1 kinesin-1 Dynactin complex V 0.8 µm/s DICs max LIS1, NudE, NuDEL Fs 1 pN, 6 pN DHC DLICs HAP1/Huntingtin Lr 1 µm DLCs Bicaudal-D family proteins Cytoplasmic dynein Vmax= Maximal velocity Fs = Stall force Lr = Length of run SnapShot: Axonal Transport Alison Twelvetrees,1,2 Adam G. -

A Platform for Brain-Wide Imaging and Reconstruction of Individual Neurons
TOOLS AND RESOURCES A platform for brain-wide imaging and reconstruction of individual neurons Michael N Economo1†, Nathan G Clack1†, Luke D Lavis1, Charles R Gerfen1,2, Karel Svoboda1, Eugene W Myers1,3*‡, Jayaram Chandrashekar1*‡ 1Janelia Research Campus, Howard Hughes Medical Institute, Ashburn, United States; 2Laboratory of Systems Neuroscience, National Institute of Mental Health, Bethesda, United States; 3Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany Abstract The structure of axonal arbors controls how signals from individual neurons are routed within the mammalian brain. However, the arbors of very few long-range projection neurons have been reconstructed in their entirety, as axons with diameters as small as 100 nm arborize in target regions dispersed over many millimeters of tissue. We introduce a platform for high-resolution, three-dimensional fluorescence imaging of complete tissue volumes that enables the visualization and reconstruction of long-range axonal arbors. This platform relies on a high-speed two-photon microscope integrated with a tissue vibratome and a suite of computational tools for large-scale image data. We demonstrate the power of this approach by reconstructing the axonal arbors of *For correspondence: myers@ multiple neurons in the motor cortex across a single mouse brain. mpi-cbg.de (EWM); DOI: 10.7554/eLife.10566.001 [email protected] (JC) †These authors contributed equally to this work Introduction ‡These authors also contributed A core goal of modern neuroscience is to map neural circuits across the entire brain, as long-range equally to this work connectivity dictates how information flows between brain regions (Bohland et al., 2009). -

A Student's Guide to Neural Circuit Tracing
fnins-13-00897 August 23, 2019 Time: 18:23 # 1 REVIEW published: 27 August 2019 doi: 10.3389/fnins.2019.00897 A Student’s Guide to Neural Circuit Tracing Christine Saleeba1,2†, Bowen Dempsey3†, Sheng Le1, Ann Goodchild1 and Simon McMullan1* 1 Neurobiology of Vital Systems Node, Faculty of Medicine and Health Sciences, Macquarie University, Sydney, NSW, Australia, 2 The School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, United Kingdom, 3 CNRS, Hindbrain Integrative Neurobiology Laboratory, Neuroscience Paris-Saclay Institute (Neuro-PSI), Université Paris-Saclay, Gif-sur-Yvette, France The mammalian nervous system is comprised of a seemingly infinitely complex network of specialized synaptic connections that coordinate the flow of information through it. The field of connectomics seeks to map the structure that underlies brain function at resolutions that range from the ultrastructural, which examines the organization of individual synapses that impinge upon a neuron, to the macroscopic, which examines gross connectivity between large brain regions. At the mesoscopic level, distant and local connections between neuronal populations are identified, Edited by: providing insights into circuit-level architecture. Although neural tract tracing techniques Vaughan G. Macefield, have been available to experimental neuroscientists for many decades, considerable Baker Heart and Diabetes Institute, methodological advances have been made in the last 20 years due to synergies between Australia the fields of molecular biology, virology, microscopy, computer science and genetics. As Reviewed by: Patrice G. Guyenet, a consequence, investigators now enjoy an unprecedented toolbox of reagents that University of Virginia, United States can be directed against selected subpopulations of neurons to identify their efferent and Eberhard Weihe, University of Marburg, Germany afferent connectomes. -

Clinical Consequences of Axonal Injury in Traumatic Brain Injury
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 1436 Clinical Consequences of Axonal Injury in Traumatic Brain Injury SAMI ABU HAMDEH ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6206 ISBN 978-91-513-0251-5 UPPSALA urn:nbn:se:uu:diva-341914 2018 Dissertation presented at Uppsala University to be publicly examined in Auditorium minus, Gustavianum, Akademigatan 3, Uppsala, Saturday, 21 April 2018 at 09:15 for the degree of Doctor of Philosophy (Faculty of Medicine). The examination will be conducted in English. Faculty examiner: Professor Andreas Unterberg MD, PhD (Department of Neurosurgery, Heidelberg University). Abstract Abu Hamdeh, S. 2018. Clinical Consequences of Axonal Injury in Traumatic Brain Injury. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 1436. 84 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-513-0251-5. Traumatic brain injury (TBI), mainly caused by road-traffic accidents and falls, is a leading cause of mortality. Survivors often display debilitating motor, sensory and cognitive symptoms, leading to reduced quality of life and a profound economic burden to society. Additionally, TBI is a risk factor for future neurodegenerative disorders including Alzheimer’s disease (AD). Commonly, TBI is categorized into focal and diffuse injuries, and based on symptom severity into mild, moderate and severe TBI. Diffuse axonal injury (DAI), biomechanically caused by rotational acceleration-deceleration forces at impact, is characterized by widespread axonal injury in superficial and deep white substance. DAI comprises a clinical challenge due to its variable course and unreliable prognostic methods. Furthermore, axonal injury may convey the link to neurodegeneration since molecules associated with neurodegenerative events aggregate in injured axons.