Endocrinology Endocrine Society
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![Computational Genome-Wide Identification of Heat Shock Protein Genes in the Bovine Genome [Version 1; Peer Review: 2 Approved, 1 Approved with Reservations]](https://docslib.b-cdn.net/cover/8283/computational-genome-wide-identification-of-heat-shock-protein-genes-in-the-bovine-genome-version-1-peer-review-2-approved-1-approved-with-reservations-88283.webp)
Computational Genome-Wide Identification of Heat Shock Protein Genes in the Bovine Genome [Version 1; Peer Review: 2 Approved, 1 Approved with Reservations]
F1000Research 2018, 7:1504 Last updated: 08 AUG 2021 RESEARCH ARTICLE Computational genome-wide identification of heat shock protein genes in the bovine genome [version 1; peer review: 2 approved, 1 approved with reservations] Oyeyemi O. Ajayi1,2, Sunday O. Peters3, Marcos De Donato2,4, Sunday O. Sowande5, Fidalis D.N. Mujibi6, Olanrewaju B. Morenikeji2,7, Bolaji N. Thomas 8, Matthew A. Adeleke 9, Ikhide G. Imumorin2,10,11 1Department of Animal Breeding and Genetics, Federal University of Agriculture, Abeokuta, Nigeria 2International Programs, College of Agriculture and Life Sciences, Cornell University, Ithaca, NY, 14853, USA 3Department of Animal Science, Berry College, Mount Berry, GA, 30149, USA 4Departamento Regional de Bioingenierias, Tecnologico de Monterrey, Escuela de Ingenieria y Ciencias, Queretaro, Mexico 5Department of Animal Production and Health, Federal University of Agriculture, Abeokuta, Nigeria 6Usomi Limited, Nairobi, Kenya 7Department of Animal Production and Health, Federal University of Technology, Akure, Nigeria 8Department of Biomedical Sciences, Rochester Institute of Technology, Rochester, NY, 14623, USA 9School of Life Sciences, University of KwaZulu-Natal, Durban, 4000, South Africa 10School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA, 30032, USA 11African Institute of Bioscience Research and Training, Ibadan, Nigeria v1 First published: 20 Sep 2018, 7:1504 Open Peer Review https://doi.org/10.12688/f1000research.16058.1 Latest published: 20 Sep 2018, 7:1504 https://doi.org/10.12688/f1000research.16058.1 Reviewer Status Invited Reviewers Abstract Background: Heat shock proteins (HSPs) are molecular chaperones 1 2 3 known to bind and sequester client proteins under stress. Methods: To identify and better understand some of these proteins, version 1 we carried out a computational genome-wide survey of the bovine 20 Sep 2018 report report report genome. -

Gene Targeting Therapies (Roy Alcalay)
Recent Developments in Gene - Targeted Therapies for Parkinson’s Disease Roy Alcalay, MD, MS Alfred and Minnie Bressler Associate Professor of Neurology Division of Movement Disorders Columbia University Medical Center Disclosures Funding: Dr. Alcalay is funded by the National Institutes of Health, the DOD, the Michael J. Fox Foundation and the Parkinson’s Foundation. Dr. Alcalay receives consultation fees from Genzyme/Sanofi, Restorbio, Janssen, and Roche. Gene Localizations Identified in PD Gene Symbol Protein Transmission Chromosome PARK1 SNCA α-synuclein AD 4q22.1 PARK2 PRKN parkin (ubiquitin ligase) AR 6q26 PARK3 ? ? AD 2p13 PARK4 SNCA triplication α-synuclein AD 4q22.1 PARK5 UCH-L1 ubiquitin C-terminal AD 4p13 hydrolase-L1 PARK6 PINK1 PTEN-induced kinase 1 AR 1p36.12 PARK7 DJ-1 DJ-1 AR 1p36.23 PARK8 LRRK2 leucine rich repeat kinase 2 AD 12q12 PARK9 ATP13A2 lysosomal ATPase AR 1p36.13 PARK10 ? ? (Iceland) AR 1p32 PARK11 GIGYF2 GRB10-interacting GYF protein 2 AD 2q37.1 PARK12 ? ? X-R Xq21-q25 PARK13 HTRA2 serine protease AD 2p13.1 PARK14 PLA2G6 phospholipase A2 (INAD) AR 22q13.1 PARK15 FBXO7 F-box only protein 7 AR 22q12.3 PARK16 ? Discovered by GWAS ? 1q32 PARK17 VPS35 vacuolar protein sorting 35 AD 16q11.2 PARK18 EIF4G1 initiation of protein synth AD 3q27.1 PARK19 DNAJC6 auxilin AR 1p31.3 PARK20 SYNJ1 synaptojanin 1 AR 21q22.11 PARK21 DNAJC13 8/RME-8 AD 3q22.1 PARK22 CHCHD2 AD 7p11.2 PARK23 VPS13C AR 15q22 Gene Localizations Identified in PD Disorder Symbol Protein Transmission Chromosome PD GBA β-glucocerebrosidase AD 1q21 SCA2 -

Identification and Characterization of TPRKB Dependency in TP53 Deficient Cancers
Identification and Characterization of TPRKB Dependency in TP53 Deficient Cancers. by Kelly Kennaley A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Molecular and Cellular Pathology) in the University of Michigan 2019 Doctoral Committee: Associate Professor Zaneta Nikolovska-Coleska, Co-Chair Adjunct Associate Professor Scott A. Tomlins, Co-Chair Associate Professor Eric R. Fearon Associate Professor Alexey I. Nesvizhskii Kelly R. Kennaley [email protected] ORCID iD: 0000-0003-2439-9020 © Kelly R. Kennaley 2019 Acknowledgements I have immeasurable gratitude for the unwavering support and guidance I received throughout my dissertation. First and foremost, I would like to thank my thesis advisor and mentor Dr. Scott Tomlins for entrusting me with a challenging, interesting, and impactful project. He taught me how to drive a project forward through set-backs, ask the important questions, and always consider the impact of my work. I’m truly appreciative for his commitment to ensuring that I would get the most from my graduate education. I am also grateful to the many members of the Tomlins lab that made it the supportive, collaborative, and educational environment that it was. I would like to give special thanks to those I’ve worked closely with on this project, particularly Dr. Moloy Goswami for his mentorship, Lei Lucy Wang, Dr. Sumin Han, and undergraduate students Bhavneet Singh, Travis Weiss, and Myles Barlow. I am also grateful for the support of my thesis committee, Dr. Eric Fearon, Dr. Alexey Nesvizhskii, and my co-mentor Dr. Zaneta Nikolovska-Coleska, who have offered guidance and critical evaluation since project inception. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association Between BMI and Adult-Onset Non- Atopic
Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association between BMI and Adult-Onset Non- Atopic Asthma Ayoung Jeong 1,2, Medea Imboden 1,2, Akram Ghantous 3, Alexei Novoloaca 3, Anne-Elie Carsin 4,5,6, Manolis Kogevinas 4,5,6, Christian Schindler 1,2, Gianfranco Lovison 7, Zdenko Herceg 3, Cyrille Cuenin 3, Roel Vermeulen 8, Deborah Jarvis 9, André F. S. Amaral 9, Florian Kronenberg 10, Paolo Vineis 11,12 and Nicole Probst-Hensch 1,2,* 1 Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; [email protected] (A.J.); [email protected] (M.I.); [email protected] (C.S.) 2 Department of Public Health, University of Basel, 4001 Basel, Switzerland 3 International Agency for Research on Cancer, 69372 Lyon, France; [email protected] (A.G.); [email protected] (A.N.); [email protected] (Z.H.); [email protected] (C.C.) 4 ISGlobal, Barcelona Institute for Global Health, 08003 Barcelona, Spain; [email protected] (A.-E.C.); [email protected] (M.K.) 5 Universitat Pompeu Fabra (UPF), 08002 Barcelona, Spain 6 CIBER Epidemiología y Salud Pública (CIBERESP), 08005 Barcelona, Spain 7 Department of Economics, Business and Statistics, University of Palermo, 90128 Palermo, Italy; [email protected] 8 Environmental Epidemiology Division, Utrecht University, Institute for Risk Assessment Sciences, 3584CM Utrecht, Netherlands; [email protected] 9 Population Health and Occupational Disease, National Heart and Lung Institute, Imperial College, SW3 6LR London, UK; [email protected] (D.J.); [email protected] (A.F.S.A.) 10 Division of Genetic Epidemiology, Medical University of Innsbruck, 6020 Innsbruck, Austria; [email protected] 11 MRC-PHE Centre for Environment and Health, School of Public Health, Imperial College London, W2 1PG London, UK; [email protected] 12 Italian Institute for Genomic Medicine (IIGM), 10126 Turin, Italy * Correspondence: [email protected]; Tel.: +41-61-284-8378 Int. -

Identification of Transcriptional Mechanisms Downstream of Nf1 Gene Defeciency in Malignant Peripheral Nerve Sheath Tumors Daochun Sun Wayne State University
Wayne State University DigitalCommons@WayneState Wayne State University Dissertations 1-1-2012 Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors Daochun Sun Wayne State University, Follow this and additional works at: http://digitalcommons.wayne.edu/oa_dissertations Recommended Citation Sun, Daochun, "Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors" (2012). Wayne State University Dissertations. Paper 558. This Open Access Dissertation is brought to you for free and open access by DigitalCommons@WayneState. It has been accepted for inclusion in Wayne State University Dissertations by an authorized administrator of DigitalCommons@WayneState. IDENTIFICATION OF TRANSCRIPTIONAL MECHANISMS DOWNSTREAM OF NF1 GENE DEFECIENCY IN MALIGNANT PERIPHERAL NERVE SHEATH TUMORS by DAOCHUN SUN DISSERTATION Submitted to the Graduate School of Wayne State University, Detroit, Michigan in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY 2012 MAJOR: MOLECULAR BIOLOGY AND GENETICS Approved by: _______________________________________ Advisor Date _______________________________________ _______________________________________ _______________________________________ © COPYRIGHT BY DAOCHUN SUN 2012 All Rights Reserved DEDICATION This work is dedicated to my parents and my wife Ze Zheng for their continuous support and understanding during the years of my education. I could not achieve my goal without them. ii ACKNOWLEDGMENTS I would like to express tremendous appreciation to my mentor, Dr. Michael Tainsky. His guidance and encouragement throughout this project made this dissertation come true. I would also like to thank my committee members, Dr. Raymond Mattingly and Dr. John Reiners Jr. for their sustained attention to this project during the monthly NF1 group meetings and committee meetings, Dr. -

The HSP70 Chaperone Machinery: J Proteins As Drivers of Functional Specificity
REVIEWS The HSP70 chaperone machinery: J proteins as drivers of functional specificity Harm H. Kampinga* and Elizabeth A. Craig‡ Abstract | Heat shock 70 kDa proteins (HSP70s) are ubiquitous molecular chaperones that function in a myriad of biological processes, modulating polypeptide folding, degradation and translocation across membranes, and protein–protein interactions. This multitude of roles is not easily reconciled with the universality of the activity of HSP70s in ATP-dependent client protein-binding and release cycles. Much of the functional diversity of the HSP70s is driven by a diverse class of cofactors: J proteins. Often, multiple J proteins function with a single HSP70. Some target HSP70 activity to clients at precise locations in cells and others bind client proteins directly, thereby delivering specific clients to HSP70 and directly determining their fate. In their native cellular environment, polypeptides are participates in such diverse cellular functions. Their constantly at risk of attaining conformations that pre- functional diversity is remarkable considering that vent them from functioning properly and/or cause them within and across species, HSP70s have high sequence to aggregate into large, potentially cytotoxic complexes. identity. They share a single biochemical activity: an Molecular chaperones guide the conformation of proteins ATP-dependent client-binding and release cycle com- throughout their lifetime, preventing their aggregation bined with client protein recognition, which is typi- by protecting interactive surfaces against non-productive cally rather promiscuous. This apparent conundrum interactions. Through such inter actions, molecular chap- is resolved by the fact that HSP70s do not work alone, erones aid in the folding of nascent proteins as they are but rather as ‘HSP70 machines’, collaborating with synthesized by ribosomes, drive protein transport across and being regulated by several cofactors. -
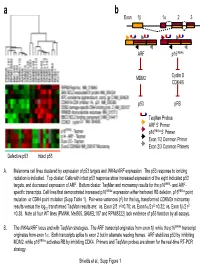
Taqman Probes ARF 5' Primer P16ink4a 5' Primer Exon 1/2
a b Exon 1β 1α 23 ARF p16INK4a MDM2 Cyclin D CDK4/6 p53 pRB TaqMan Probes ARF 5’ Primer p16INK4a 5’ Primer Exon 1/2 Common Primer Exon 2/3 Common Primers Defective p53 Intact p53 A. Melanoma cell lines clustered by expression of p53 targets and INK4a/ARF expression. The p53 response to ionizing radiation is indicated. Top cluster: Cells with intact p53 response show increased expression of the eight indicated p53 targets, and decreased expression of ARF. Bottom cluster: TaqMan and microarray results for the p16INK4- and ARF- specific transcripts. Cell lines that demonstrated increased p16INK4a expression either harbored RB deletion, p16INK4a point 2 mutation or CDK4 point mutation (Supp Table 1). Pair-wise variances (r ) for the log2 transformed CDKN2a microarray 2 2 2 results versus the log10 transformed TaqMan results are: vs. Exon 2/3 r =0.78; vs. Exon1α/2 r =0.32; vs. Exon 1β/2 r =0.38. Note all four WT lines (PMWK, Mel505, SKMEL187 and RPMI8322) lack evidence of p53 function by all assays. B. The INK4a/ARF locus and with TaqMan strategies. The ARF transcript originates from exon 1β while the p16INK4a transcript originates from exon 1α. Both transcripts splice to exon 2 but in alternate reading frames. ARF stabilizes p53 by inhibiting MDM2, while p16INK4a activates RB by inhibiting CDK4. Primers and TaqMan probes are shown for the real-time RT-PCR strategy. Shields et al., Supp Figure 1 SKMEL 28 U01 24h SKMEL WM2664 U01 48h WM2664 U01 24h 24 U01 48h SKMEL 24 U01 24h SKMEL 24 Untreated SKMEL 24 DMSO 48h SKMEL 24 DMSO 24h SKMEL WM2664 DMSO 24h WM2664 DMSO 48h WM2644 Untreated 28 Untreated SKMEL 28 DMSO 48h SKMEL 28 DMSO 24h SKMEL -3.00 -2.00 -1.00 0.00 1.00 2.00 3.00 relative to median expression Genes decreased by UO1 (863) HIF1A Hypoxia-inducible factor 1, alpha subunit NM_001530 RBBP8 Retinoblastoma binding protein 8 NM_002894 Homo sapiens, clone IMAGE:4337652, mRNA BC018676 EIF4EBP1 Eukaryotic translation initiation factor 4E binding protein EXOSC8 Exosome component 8 NM_181503 ENST00000321524 MCM7 MCM7 minichromosome maintenance deficient 7 (S. -

Prognostic and Functional Significant of Heat Shock Proteins (Hsps)
biology Article Prognostic and Functional Significant of Heat Shock Proteins (HSPs) in Breast Cancer Unveiled by Multi-Omics Approaches Miriam Buttacavoli 1,†, Gianluca Di Cara 1,†, Cesare D’Amico 1, Fabiana Geraci 1 , Ida Pucci-Minafra 2, Salvatore Feo 1 and Patrizia Cancemi 1,2,* 1 Department of Biological Chemical and Pharmaceutical Sciences and Technologies (STEBICEF), University of Palermo, 90128 Palermo, Italy; [email protected] (M.B.); [email protected] (G.D.C.); [email protected] (C.D.); [email protected] (F.G.); [email protected] (S.F.) 2 Experimental Center of Onco Biology (COBS), 90145 Palermo, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-091-2389-7330 † These authors contributed equally to this work. Simple Summary: In this study, we investigated the expression pattern and prognostic significance of the heat shock proteins (HSPs) family members in breast cancer (BC) by using several bioinfor- matics tools and proteomics investigations. Our results demonstrated that, collectively, HSPs were deregulated in BC, acting as both oncogene and onco-suppressor genes. In particular, two different HSP-clusters were significantly associated with a poor or good prognosis. Interestingly, the HSPs deregulation impacted gene expression and miRNAs regulation that, in turn, affected important bio- logical pathways involved in cell cycle, DNA replication, and receptors-mediated signaling. Finally, the proteomic identification of several HSPs members and isoforms revealed much more complexity Citation: Buttacavoli, M.; Di Cara, of HSPs roles in BC and showed that their expression is quite variable among patients. In conclusion, G.; D’Amico, C.; Geraci, F.; we elaborated two panels of HSPs that could be further explored as potential biomarkers for BC Pucci-Minafra, I.; Feo, S.; Cancemi, P. -

Characterization of the Dual Functional Effects of Heat Shock Proteins (Hsps
Zhang et al. Genome Medicine (2020) 12:101 https://doi.org/10.1186/s13073-020-00795-6 RESEARCH Open Access Characterization of the dual functional effects of heat shock proteins (HSPs) in cancer hallmarks to aid development of HSP inhibitors Zhao Zhang1†, Ji Jing2†, Youqiong Ye1, Zhiao Chen1, Ying Jing1, Shengli Li1, Wei Hong1, Hang Ruan1, Yaoming Liu1, Qingsong Hu3, Jun Wang4, Wenbo Li1, Chunru Lin3, Lixia Diao5*, Yubin Zhou2* and Leng Han1* Abstract Background: Heat shock proteins (HSPs), a representative family of chaperone genes, play crucial roles in malignant progression and are pursued as attractive anti-cancer therapeutic targets. Despite tremendous efforts to develop anti-cancer drugs based on HSPs, no HSP inhibitors have thus far reached the milestone of FDA approval. There remains an unmet need to further understand the functional roles of HSPs in cancer. Methods: We constructed the network for HSPs across ~ 10,000 tumor samples from The Cancer Genome Atlas (TCGA) and ~ 10,000 normal samples from Genotype-Tissue Expression (GTEx), and compared the network disruption between tumor and normal samples. We then examined the associations between HSPs and cancer hallmarks and validated these associations from multiple independent high-throughput functional screens, including Project Achilles and DRIVE. Finally, we experimentally characterized the dual function effects of HSPs in tumor proliferation and metastasis. Results: We comprehensively analyzed the HSP expression landscape across multiple human cancers and revealed a global disruption of the co-expression network for HSPs. Through analyzing HSP expression alteration and its association with tumor proliferation and metastasis, we revealed dual functional effects of HSPs, in that they can simultaneously influence proliferation and metastasis in opposite directions. -

RT² Profiler PCR Array (Rotor-Gene® Format) Mouse Heat Shock Proteins & Chaperones
RT² Profiler PCR Array (Rotor-Gene® Format) Mouse Heat Shock Proteins & Chaperones Cat. no. 330231 PAMM-076ZR For pathway expression analysis Format For use with the following real-time cyclers RT² Profiler PCR Array, Rotor-Gene Q, other Rotor-Gene cyclers Format R Description The Mouse Heat Shock Proteins & Chaperones RT² Profiler PCR Array profiles the expression of 84 Heat Shock Protein genes that regulate protein folding. Heat shock proteins (HSPs, or molecular chaperones) are important components of cellular networks. HSPs can either aid in the folding and maintenance of newly translated proteins or they can lead to the degradation of misfolded and destabilized proteins. Included on this PCR Array are HSP90 (81 to 99 kD), HSP70 (65 to 80 kD), HSP60 (55 to 64 kD), HSP40 (35 to 54 kD), small HSPs (=34 kD) and other chaperone cofactors that are directly involved in the response to unfolded/misfolded proteins or that are involved in protein folding in general. Using real-time PCR, you can easily and reliably analyze expression of a focused panel of heat shock proteins and chaperones with this array. For further details, consult the RT² Profiler PCR Array Handbook. Shipping and storage RT² Profiler PCR Arrays in the Rotor-Gene format are shipped at ambient temperature, on dry ice, or blue ice packs depending on destination and accompanying products. For long term storage, keep plates at –20°C. Note: Ensure that you have the correct RT² Profiler PCR Array format for your real-time cycler (see table above). Note: Open the package and store the products appropriately immediately on receipt. -

Genome-Wide Profiling of Druggable Active Tumor Defense Mechanisms to Enhance Cancer Immunotherapy
bioRxiv preprint doi: https://doi.org/10.1101/843185; this version posted November 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Genome-wide profiling of druggable active tumor defense mechanisms to enhance cancer immunotherapy Rigel J. Kishton1,2,*,#, Shashank J. Patel1,2,†,*, Suman K. Vodnala1,2, Amy E. Decker3, Yogin Patel1,2, Madhusudhanan Sukumar1,2, Tori N. Yamamoto1,2,4, Zhiya Yu1,2, Michelle Ji1,2, Amanda N. Henning1,2, Devikala Gurusamy1,2, Douglas C. Palmer1,2, Winifred Lo1, Anna Pasetto1, Parisa Malekzadeh1, Drew C. Deniger1, Kris C. Wood3, Neville E. Sanjana5,6, Nicholas P. Restifo1,2, #, § 1Surgery Branch, Center for Cancer Research, National Cancer Institute, Bethesda, MD 20892, USA 2Center for Cell-Based Therapy, National Cancer Institute, Bethesda, MD 20892, USA 3Department of Pharmacology & Cancer Biology, Duke University School of Medicine, Durham, NC, USA 4Immunology Graduate Group, University of Pennsylvania, Philadelphia, PA 19104, USA 5New York Genome Center, New York, NY 10013 USA 6Department of Biology, New York University, New York, NY 10003, USA *These authors contributed equally to this work. †Present address: NextCure Inc., Beltsville, MD 20705, USA §Present address: Lyell Immunopharma, South San Francisco, CA 94080, USA #Corresponding authors. NPR: [email protected]. RJK: [email protected]. bioRxiv preprint doi: https://doi.org/10.1101/843185; this version posted November 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission.