MEMOTE for Standardized Genome-Scale Metabolic Model Testing
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Men's All-Time World Performers-Performances Rankings
Men’s All-Time World Performers-Performances Rankings Page 1 of 127 50 METER BACKSTROKE Top 2660 Performances 24.04** Liam Tancock, GBR 13th World Championships Rome 08-02-09 (Reaction Time: +0.60. (Note: Great Britain’s first male backstroke gold-medalist [50, 100, 200]. Tancock’s first international gold/second world- record. (Note: bronze medalist [2005, Montreal; ’07, Melbourne]) 24.07*# Camille Lacourt, FRA XXX European Championships Budapest 08-12-10 (Reaction Time: +0.74. (Nore: also clocked European-record/history’second-fastest 100 back en route to gold several days earlieir [52.11]) 24.08sf1 Tancock 13th World Championships Rome 08-01-09 (Reaction Time: +0.57) 24.23 Lacourt 16th World Championships Kazan 08-09-15 (Reaction Time: +0.68, gold medalist) 24.24a Junya Koga, JPN 13th World Championships Rome 08-02-09 (Reaction Time: +0.50. (Note: won 100 back gold in an Asian-record 52.26 clocking several days earlier.) 24.27sf2 Lacourt 16th World Championships Kazan 08-08-15 (Reaction Time: +0.69) 24.28 Koga 17th Asian Games Incheon 09-21-14 (Reaction Time: +0.52 [fastest of race]. (Note: Games record, Koga’s third-consecutive gold/record. Won @ Doha in 2K6 [25.40]; Guangzhou, 2K10 [25.08]) 24.29sf2 Koga 13th World Championships Rome 08-01-09 (Reaction Time: +0.48) 24.30sf1 Lacourt XXX European Championships Budapest 08-11-10 (Reaction Time: +0.71) 24.33* Randall Bal, USA/Stanford Eindhoven Swim Cup Eindhoven 12-05-08 (Reaction Time: +0.66) 24.34* Gerhard Zandberg, RSA/Arizona 13th World Championshps Rome 08-02-09 (Note: African record.) 24.36 Lacourt FRA Nationals/WCTs Strasbouug 03-27-11 (Note: French Open-“All Comers” record.) 24.37 Lacourt FRA Nats./Euro. -

EUROPEAN SHORT COURSE Swimming Championships Finalists
EUROPEAN SHORT COURSE Swimming Championships Finalists 20191 Some of the stars of the European Short Course Championships in Copenhagen- top row, left Kirill Prigoda (Russia), centre, Adam Peaty (Great Britain), right, Ruta Meilutyte (Lithuania); middle row, left, Radoslaw Kawecki (Poland), centre, Maxence Orange (France), right, Fanny Lecluyse (Belgium); bottom, left, Andri Govorov (Ukraine), centre, Sarah Koehler (Germany) and Julia Hassler (Liechtenstein), right, Matteo Rivolta (Italy) (Photos: Giorgio Scala & Andrea Staccioli, Deepbluemedia/Insidefoto) 2 European Short Course Swimming Championships Finalists Contents European Sprint Results - Men 4 European Sprint Results - Women 7 European Short Course Championship Venues 10 Short Course Results - Men 11 Short Course Results - Women 88 Short Course Results - Mixed 164 European Sprint Championships Medals Tables - by country 167 European Sprint Championships Medals Tables - by event 169 European Short Course Medals Tables - by country 172 European Short Course Medals Tables - by event 178 European Short Course Leading Medallists - all time 191 Please note that, unless stated otherwise, the photos in this book were taken at the 2017 European Short Course Championships in Copenhagen 3 European Sprints Results 1991 to 1994 This book is in two sections. The first The first European Sprints were held section deals with the European Sprint between December 6th and 8th 1991 at Championships held between 1991 and Gelsenkirchen, Germany when the city 1994; the second, with the European agreed to organise the event with only Short Course Championships from 1996 to four months notice. The first European the present time. The tables of individual Short Course Championships in Rostock medals and event medal tables at the end in 1996 saw a significant expansion with of this publication, therefore, treats them some 36 events. -

Giorno 3. Panziera E Paltrinieri D’
Giorno 3. Panziera e Paltrinieri d’Oro. Bianchi d’Argento. written by Redazione | 6 Dicembre 2019 Oro Panziera con record italiano. Oro Paltrinieri. Argento Bianchi con primato nazionale. RECAP FINALI Photo Giorgio Scala / Deepbluemedia / Insidefoto Sua “Maestà” Gregorio Paltrinieri Con un improvviso cambio di ritmo portato ai settecento metri il fuoriclasse carpigiano si porta in testa alla gara fino al touch finale e si riprende il trono continentale dei 1500 stile libero. (14,17.14) davanti al temibile norvegese Henrik Christiansen (14.18.15), bronzo al francese David Aubry, resta fuori dal podio l’ucraino Mykhaylo Romanchuk che a metà gara sembra addirittura rinunciare alla lotta per le medaglie. Chiude la finale in ottava posizione l’azzurro Domenico Acerenza (14.51.59). “Sono contento perchè torno a vincere un 1500 dopo tanto tempo. All’inizio ho visto l’ucraino che era partito forte e sono rimasto in galleggiamento. Poi a metà gara ho notato che ha rallentato e allora ho inziato la mia progressione per fortuna vincente. Oggi non avevo le stesse sensazioni di ieri e allora mi sono tenuto coperto, passando un po’ più calmo per poi aprire il gas. Erano passati troppi anni dalla vittoria. Non mi sentivo benissimo e allora ho cercato di non strafare e di andare in progressione. La forma è cambiata dalla batteria alla finale e quindi bisognerà lavorare perchè per fare questo tempo ho dato tutto”. Gregorio Paltrinieri (Federnuoto) Il video dei 1500 stile libero (Raisport) Podio 1500 sl uomini 1) Gregorio Paltrinieri (ITA) 14.17.14 2) Henrik Christiansen (NOR) 14.18.15 3) David Aubry (FRA) 14.25.66 8) Domenico Acerenza (ITA) 14.51.49 Photo Giorgio Scala / Deepbluemedia / Insidefoto Finalmente Margherita Panziera. -

CHG Library Book List
CHG Library Book List (Belgium), M. r. d. a. e. d. h. (1967). Galerie de l'Asie antérieure et de l'Iran anciens [des] Musées royaux d'art et d'histoire, Bruxelles, Musées royaux d'art et dʹhistoire, Parc du Cinquantenaire, 1967. Galerie de l'Asie antérieure et de l'Iran anciens [des] Musées royaux d'art et d'histoire by Musées royaux d'art et d'histoire (Belgium) (1967) (Director), T. P. F. H. (1968). The Metropolitan Museum of Art Bulletin: Volume XXVI, Number 5. New York: Metropolitan Museum of Art (January, 1968). The Metropolitan Museum of Art Bulletin: Volume XXVI, Number 5 by Thomas P.F. Hoving (1968) (Director), T. P. F. H. (1973). The Metropolitan Museum of Art Bulletin: Volume XXXI, Number 3. New York: Metropolitan Museum of Art (Ed.), A. B. S. (2002). Persephone. U.S.A/ Cambridge, President and Fellows of Harvard College Puritan Press, Inc. (Ed.), A. D. (2005). From Byzantium to Modern Greece: Hellenic Art in Adversity, 1453-1830. /Benaki Museum. Athens, Alexander S. Onassis Public Benefit Foundation. (Ed.), B. B. R. (2000). Christian VIII: The National Museum: Antiquities, Coins, Medals. Copenhagen, The National Museum of Denmark. (Ed.), J. I. (1999). Interviews with Ali Pacha of Joanina; in the autumn of 1812; with some particulars of Epirus, and the Albanians of the present day (Peter Oluf Brondsted). Athens, The Danish Institute at Athens. (Ed.), K. D. (1988). Antalya Museum. İstanbul, T.C. Kültür ve Turizm Bakanlığı Döner Sermaye İşletmeleri Merkez Müdürlüğü/ Ankara. (ed.), M. N. B. (Ocak- Nisan 2010). "Arkeoloji ve sanat. (Journal of Archaeology and Art): Ölümünün 100.Yıldönümünde Osman Hamdi Bey ve Kazıları." Arkeoloji Ve Sanat 133. -

Natalie Coughlin’S Success in the January 2014 - Volume 55 - No
MIXING UP YOUR TRAINING A YEAR IN REVIEW: 2013’s TOP SWIMMING STORIES A LOOK +INTO 2014 THE GLORY OF FOOD NATALIE COUGHLIN’S SUCCESS IN THE JANUARY 2014 - VOLUME 55 - NO. 01 KITCHEN $3.95 PHELPS CLEARS SURVIVE & FIRST HURDLE THRIVE: IN COMEBACK CRAIG DIETZ LEARN MORE AT SWIMMINGWORLD.COM AMERICA’S GOLDEN GIRL: CoughlinNatalie SPECIAL NEW YEAR OFFER: CHECK OUT PAGE 65 FOR SWIMMING WORLD’S NEW YEAR SPECIAL! JANUARY.indd 1 12/22/13 5:34 PM JANUARY.indd 2 12/22/13 5:34 PM JANUARY.indd 3 12/22/13 5:35 PM JANUARY.indd 4 12/22/13 5:35 PM JANUARY.indd 5 12/22/13 5:35 PM 029 Food, Glorious Food! by Natalie Coughlin Natalie Coughlin taught herself how 2014 to cook when she was in college...and JAN she absolutely loves it! 031 Take Home Points by Dr. G. John Mullen In a partnership with Swimming World 020 U Magazine, Swimming Science provides “Take Home Points” from in-depth 045 Q&A with Coach swimming research. Jason Turcotte ARY by Michael J. Stott FEATURES 032 Yoga for Swimmers by Steve Krojniewski 047 How They Train Gunnar 010 Swimming Stories of 2013 Yoga can help swimmers strengthen Bentz and Kylie Stewart 010 by Jason Marsteller their core, stretch the pectorals, open by Michael J. Stott the shoulders and add focus for peak 014 Lessons with the Legends: performance. 048 Survive and Thrive: Dick Hannula Finding A Way by Michael J. Stott 034 Goldminds: Swim to Win! by Shoshanna Rutemiller by Wayne Goldsmith Despite not having any arms or legs, 016 Strategic Pacing in Commitment—the decision to turn 39-year-old Craig Dietz is using his Distance Events (Part II) thoughts into actions—is the key to success in open water swimming to by Michael J. -
Helsinki 2010
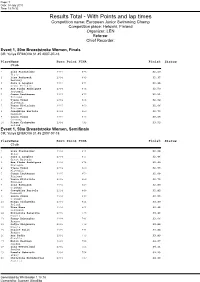
Page: 1 Date: 18 July 2010 Time: 19:18:32 Results Total - With Points and lap times Competition name: European Junior Swimming Champ Competition place: Helsinki, Finland Organizer: LEN Referee: Chief Recorder: Event 1, 50m Breaststroke Women, Finals CR: Yuliya EFIMOVA 31.45 2007-07-18 Place Name Born Point FINA Finish Status Club 1 Lisa Fissneider 1994 898 32.20 Italy 2 Lina Rathsack 1994 883 32.37 Germany 3 Sara L Lougher 1994 874 32.48 Great Britain 4 Ana Pinho Rodrigues 1994 873 32.50 Portugal 5 Jenna Laukkanen 1995 872 32.51 Finland 6 Tjasa Vozel 1994 863 32.62 Slovenia 7 Veera Kivirinta 1995 860 32.66 Finland 8 Josephina Bartela 1994 850 32.79 Denmark 9 Laura Simon 1994 816 33.24 Germany 10 Kinga Cichowska 1994 795 33.53 Poland Event 1, 50m Breaststroke Women, Semifinals CR: Yuliya EFIMOVA 31.45 2007-07-18 Place Name Born Point FINA Finish Status Club 1 Lisa Fissneider 1994 891 32.28 Italy 2 Sara L Lougher 1994 876 32.46 Great Britain 3 Ana Pinho Rodrigues 1994 874 32.49 Portugal 4 Tjasa Vozel 1994 860 32.66 Slovenia 5 Jenna Laukkanen 1995 859 32.68 Finland 6 Veera Kivirinta 1995 852 32.76 Finland 7 Lina Rathsack 1994 849 32.80 Germany 8 Josephina Bartela 1994 848 32.82 Denmark 9 Laura Simon 1994 832 33.03 Germany 10 Kinga Cichowska 1994 811 33.30 Poland 11 Tina Meza 1994 801 33.45 Slovenia 12 Elizaveta Bazarova 1995 798 33.49 Russia 13 Fanny Deberghes 1994 786 33.66 France 14 Sofya Zhigunova 1995 773 33.84 Russia 15 Claire Polit 1995 771 33.88 France 16 Ana Radic 1994 770 33.89 Croatia 17 Malin Westman 1995 758 34.07 Sweden 18 Lina -

Women's 1500M Free (Heat A) Final
Women's 1500m Free (Heat A) Final 2019 TYR Pro Swim Richmond | April 13, 2019 World Record: 15:20.48 Katie Ledecky, USA, Indianapolis, USA, 5/16/2018 (28.09, 58.50, 1:29.26, 2:00.25, 2:31.11, 3:02.50, 3:33.71, 4:04.88, 4:35.86, 5:06.82, 5:37.52, 6:08.29, 6:39.28, 7:10.13, 7:40.88, 8:11.70, 8:42.52, 9:13.20, 9:43.96, 10:14.83, 10:45.43, 11:16.15, 11:47.05, 12:17.94, 12:48.62, 13:19.43, 13:50.27, 14:20.71, 14:51.26, 15:20.48) American Record: 15:20.48 Katie Ledecky, USA, Indianapolis, USA, 5/16/2018 (28.09, 58.50, 1:29.26, 2:00.25, 2:31.11, 3:02.50, 3:33.71, 4:04.88, 4:35.86, 5:06.82, 5:37.52, 6:08.29, 6:39.28, 7:10.13, 7:40.88, 8:11.70, 8:42.52, 9:13.20, 9:43.96, 10:14.83, 10:45.43, 11:16.15, 11:47.05, 12:17.94, 12:48.62, 13:19.43, 13:50.27, 14:20.71, 14:51.26, 15:20.48) U.S. Open Record: 15:20.48 Katie Ledecky, USA, Indianapolis, USA, 5/16/2018 (28.09, 58.50, 1:29.26, 2:00.25, 2:31.11, 3:02.50, 3:33.71, 4:04.88, 4:35.86, 5:06.82, 5:37.52, 6:08.29, 6:39.28, 7:10.13, 7:40.88, 8:11.70, 8:42.52, 9:13.20, 9:43.96, 10:14.83, 10:45.43, 11:16.15, 11:47.05, 12:17.94, 1 Daniella Van Den Berg National Record: 17:07.88 ARU Daniella Van Den Berg Top 10 Performers This Year (2019 Calendar Year) 2018 1. -

SWIM MART ˉUVW H[SHULHQFH LQ DQG DURXQG WKH ZDWHU in a Structured Environment Is a Positive One
MEET YOUR MOTIVATION > ONWARD & UPWARD INSIDE COACH PAUL GRAHAM’S SUCCESS FEBRUARY 2013 - VOLUME 54 - NO. 2 DISCOVER YOUR SOURCE OF MOTIVATION + TIPS TO STAY ON 4TRACK KATINKA ON THE LEARN THE “DO”-NUTS AND* BOLTS OF MOVE... SWIMMING THOUGHTS FROM THE FRONT LINE > DAGNY KNUTSON’S AND GEMMA SPOFFORTH’S BATTLES EVEN NATURE’S LAWS WERE MEANT TO BE BROKEN NIKE FLEX LT @V\KVU»[OH]L[VILHZJPLU[PZ[[VRUV^ [OLJVTWL[P[P]LHK]HU[HNLVM[OPZYL]VS\[PVU PULSP[LWLYMVYTHUJLZ\P[Z*YLH[LKMYVT VULWPLJLVMIVUKLKMHIYPJ[OL5PRL -SL_3;OLSWZKLZ[YV`KYHNHUKSL[Z`V\ ZSPJL[OYV\NO[OL^H[LYPUHJVTWSL[LS` Z[YLHTSPULKZ[HIPSPaPUNUVZLHTZ\P[ >P[OIVUKLKZLHTZHUKHJVTWYLZZLK MP[[VOLSWRLLW^H[LYV\[P[»Z[OLVW[PT\T VW[PVUPUSPNO[^LPNO[Z^PT[LJOUVSVN` THE ALL NEW PINK LEOPARD WING BACK THEFINALS.COM FEATURES 030 The Role of Film in Swim Training 2013 012 A Meteoric Rise by Michael J. Stott Inside by Shoshanna Rutemiller FEBRUARY In the first of two parts, Swimming Katinka Hosszu has come a long way World Magazine examines some since representing Hungary in her first history and basics about videoing Olympics as a 15-year-old in 2004. swimmers. Two more Olympic Games later—as well as becoming a multiple world 032 USSSA: The “Do”-Nuts and champion and the top female swimmer Bolts of Swimming at the recent World Cup Circuit with 39 by Tracy Laman individual gold medals—she now has her sights set on even more hardware. As learn-to-swim instructors, it is their job to make sure a child’s first 012 017 Thoughts From The Front experience in and around the water Line: Battling With in a structured environment is a Disordered Eating positive one. -

Swimming World Biweekly on the Cover - Olivia Smoliga by Peter H
make your move. NICK GRAINGER ARIANNA VANDERPOOL-WALLACE ROBBIE RENWICK TOM LAXTON 2015 WORLD CHAMPION 2008, 2012 OLYMPIAN 2015 WORLD CHAMPION BRITISH NATIONAL CHAMPION CHALLENGER FUSE ONYX VAPOR Fully Customizable UV Protective Diamond Flex Lightweight Textile Textile Textile Compressed Textile Versatile Fit Versatile Fit Engineered Fit Structured Fit Flexible Flexible Complete Selective Compression Compression Double Panel Double Panel Compression Compression 2016 TECHNICAL RACING SUITS #makeyourmove2016 To learn more, contact your local dealer or visit FINISinc.com ISSUE #2 OF MARCH 2016 FEATURES BIWEEKLy 006 WOMEN’S NCAA DIVISION I 028 HISTORIC SURPRISES AT U.S. OLYMPIC CHAMPIONSHIP FINALS RECAP TRIALS: JEFF FARRELL by Jason Marsteller by Annie Grevers The full recap from Atlanta as the ladies In 1960, sportswriters had dubbed Jeff Farrell “the from Georgia dazzle the home crowd. fastest swimmer in the world.” However, less than a week before the U.S. Olympic Trials, he underwent an emergency appendectomy... PUBLISHING, CIRCULATION AND 012 JACK BAUERLE, LILLY KING ACCOUNTING OFFICE HONORED BY CSCAA GILES SMITH MOVES TO 4TH IN WORLD IN by Jason Marsteller 032 www.SwimmingWorldMagazine.com The College Swimming Coaches Association of America 100 FLY AT FEDERAL WAY SECTIONALS by Jason Marsteller Chairman of the Board, President - Richard Deal (CSCAA) announced award recipients from the 2016 [email protected] NCAA Division I Women’s Swimming and Diving Phoenix’s Giles Smith crushed the Olympic Trials cut Championships. in the men’s 100-meter fly. His 52.10 shot him to fourth Publisher, CEO - Brent T. Rutemiller in the world behind only Laszlo Cseh (51.40), Michael [email protected] 013 NCAA DI CHAMPIONSHIP PHOTO GALLERY Phelps (51.94) and Zhuhao Li (51.97). -

Supplemental Digital Content
Supplemental Digital Content Study Protocol: Longitudinal course of depressive, anxiety and post-traumatic stress disorder symptoms after heart surgery Review question What is the longitudinal course of depressive, anxiety, and post-traumatic stress-disorder (PTSD) symptoms in cardiac patients undergoing heart surgery? Searches PubMed and Scopus, up to 07/31/2019, search key: ("implantable cardiac defibrillator" OR "cardiac operation" OR "heart surgery" OR "cardiac surgery" OR "coronary artery bypass" OR "open heart surgery" OR "valve surgery" OR “heart transplant*”) AND ((("depression" OR "depressive") AND "episode") OR "major depressive disorder" OR "anxiety" OR "PTSD" OR "post-traumatic stress disorder" OR "acute stress- reaction" OR "quality of life"). The Preferred Reporting Items for Systematic RevieWs and Meta-analyses (PRISMA) recommendations will be used to guide this meta-analysis. (1) Inclusion criteria Cohort studies measuring depressive, anxiety, or PTSD symptoms with validated psychometric tools, before and at least 1 month after heart surgery. Exclusion criteria Article type different from cohort study (interventional, revieW article, meta-analysis, editorial, conference abstract, case report). Language other than English. Publication date antecedent to 1980. Population Cardiac patients scheduled for heart surgery. Exposure Cardiac surgery, whether first time or more. Comparator Post-surgical depressive, anxiety, and PTSD symptoms, whether reported as mean scores or rates by the authors, will be compared with the pre-surgical levels in the same study population. Primary outcome Primary outcome Will be the pre-post change in depressive, anxiety, and PTSD symptoms folloWing surgery, measured as standardized effect size. Secondary Outcomes Secondary outcomes Will be: 1) the durability of mental health symptom changes; 2) the correlation betWeen baseline demographic and clinical characteristics and improvement in mental health scores. -

ISL 2020 Team
NY BREAKERS LA CURRENT CALI CONDORS DC TRIDENT ENERGY STANDARD IRON LONDON ROAR AQUA CENTURIONS TORONTO TITANS TOKYO FROG KINGS MEN COUNTRY MEN COUNTRY MEN COUNTRY MEN COUNTRY MEN COUNTRY MEN COUNTRY MEN COUNTRY MEN COUNTRY MEN COUNTRY MEN COUNTRY 1 Michael Andrew USA 1 Dylan Carter TTO 1 Townley Haas USA 1 Tristan Hollard AUS 1 Chad Le Clos RSA 1 Robert Glinta ROU 1 Adam Peaty GBR 1 Travis Mahoney AUS 1 Michael Chadwick USA 1 Daiya Seto JPN 2 Marco Koch GER 2 Felipe Silva BRA 2 Tate Jackson USA 2 Conner McHugh USA 2 Florent Manaudou FRA 2 Maxim Lobanovszkij HUN 2 Kyle Chalmers AUS 2 Fabio Scozzoli ITA 2 Mack Darragh CAN 2 Shinri Shioura JPN 3 Chris Reid RSA 3 Fernando Scheffer BRA 3 Kacper Majchrzak POL 3 Matheus Santana BRA 3 Max Litchfield GBR 3 Kristof Milak HUN 3 James Guy GBR 3 Sebastian Szabo HUN 3 Sergey Fesikov RUS 3 Markus Thormeyer CAN 4 Brendon Smith AUS 4 Kristian Gkolomeev GRE 4 Radoslaw Kawecki POL 4 Zach Apple USA 4 Andrius Sidlauskas LTU 4 Ross Murdoch GBR 4 Tom Dean GBR 4 Alessandro Miressi ITA 4 Andriy Govorov UKR 4 Vlad Morozov RUS 5 Brandon Almeida BRA 5 Andrew Seliskar USA 5 Mark Szaranek GBR 5 Zach Harting USA 5 Evgeny Rylov RUS 5 Yakov Toumarkin ISR 5 Elijah Winnington AUS 5 Matteo Rivolta ITA 5 Brent Hayden CAN 5 Yasuhiro Koseki JPN 6 Felix Aubock AUT 6 Santiago Grassi ARG 6 Clyde Lewis AUS 6 Zane Grothe USA 6 Felipe Lima BRA 6 Guilherme Basseto BRA 6 Alex Graham AUS 6 Nicolo Martinenghi ITA 6 Yuri Kisil CAN 6 Katsuhiro Matsumoto JPN 7 Jack McLoughlin AUS 7 Tom Shields USA 7 Justin Ress USA 7 Robert Howard USA -

Giorno 5. ORO Quadarella, Miressi, Di Liddo E Veloce Femminile D’
Giorno 5. ORO Quadarella, Miressi, Di Liddo e veloce femminile d’argento, Carraro di bronzo written by Redazione | 8 Dicembre 2019 Con la quinta giornata di gare si conclude a Glasgow la ventesima edizione dei Campionati Europei di nuoto in vasca corta. Dalla giornata odierna arrivano per noi altre soddisfazioni, 1 Oro, 3 Argenti, 1 Bronzo, 3 nuovi primati nazionali e 2 medaglie di legno. Segue il recap della quinta giornata di gare. RECAP FINALI DELL’ULTIMA GIORNATA Photo Giorgio Scala / Deepbluemedia / Insidefoto Doppietta d’oro continentale per la nostra campionessa del mondoSimona Quadarella , la romana conquista il titolo europeo dei 400 stile libero dopo aver vinto quello sulla doppia distanza ad inizio rassegna. L’atleta diChristian Minotti dimostra una volta ancora la straordinaria capacità di leggere ed interpretare la gara con una percezione delle avversarie fuori dal comune, il poderoso cambio di ritmo imposto dall’azzurra ai settecento metri di gara le ha permesso di scrollarsi di dosso le avversarie per aver quel minimo di luce necessaria per chiudere la prova davanti alla temibile tedesca Isabel Gose che ha condotto la prima parte di gara(4.00.01), segue per il bronzo l’unghereseAnya Kesely (4.00.04), la nostra portacolori è l’unica a coprire la distanza sotto i 4 minuti (3.59.75). Photo Giorgio Scala / Deepbluemedia / Insidefoto Sono arrivata sul blocchetto con la consapevolezza di volere conquistare l’oro. Sono stati giorni un po’ difficili, anche perché abbiamo preparato le gare pure pensando agli assoluti; poi si è accesa una lampadina. Vedere 3’59 è meraviglioso; ci provavo già dalla scorsa stagione.