1 a Gene Implicated in Activation of Retinoic Acid Receptor Targets Is A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Construction of a Natural Panel of 11P11.2 Deletions and Further Delineation of the Critical Region Involved in Potocki–Shaffer Syndrome
European Journal of Human Genetics (2005) 13, 528–540 & 2005 Nature Publishing Group All rights reserved 1018-4813/05 $30.00 www.nature.com/ejhg ARTICLE Construction of a natural panel of 11p11.2 deletions and further delineation of the critical region involved in Potocki–Shaffer syndrome Keiko Wakui1,12, Giuliana Gregato2,3,12, Blake C Ballif2, Caron D Glotzbach2,4, Kristen A Bailey2,4, Pao-Lin Kuo5, Whui-Chen Sue6, Leslie J Sheffield7, Mira Irons8, Enrique G Gomez9, Jacqueline T Hecht10, Lorraine Potocki1,11 and Lisa G Shaffer*,2,4 1Department of Molecular & Human Genetics, Baylor College of Medicine, Houston, TX, USA; 2Health Research and Education Center, Washington State University, Spokane, WA, USA; 3Dip. Patologia Umana ed Ereditaria, Sez. Biologia Generale e Genetica Medica, Universita` degli Studi di Pavia, Italy; 4Sacred Heart Medical Center, Spokane, WA, USA; 5Department of Obstetrics and Gynecology, National Cheng-Kung University Medical College, Taiwan; 6Department of Pediatrics, Taipei Municipal Women and Children’s Hospital, Taiwan; 7Genetic Health Services Victoria, Murdoch Children’s Research Institute, Department of Paediatrics, University of Melbourne, Victoria, Australia; 8Division of Genetics, Department of Medicine, Children’s Hospital, Harvard Medical School, Boston, MA, USA; 9Area de Gene´tica, Centro de Desarrollo Infantil y Departamento de Pediatrı´a Hospital Materno Infantil-Hospital Regional Universitario ‘Infanta Cristina’, Badajoz, Spain; 10Department of Pediatrics, University of Texas Medical School at Houston, TX, USA; 11Texas Children’s Hospital, Houston, TX, USA Potocki–Shaffer syndrome (PSS) is a contiguous gene deletion syndrome that results from haploinsufficiency of at least two genes within the short arm of chromosome 11[del(11)(p11.2p12)]. -

CREB-Dependent Transcription in Astrocytes: Signalling Pathways, Gene Profiles and Neuroprotective Role in Brain Injury
CREB-dependent transcription in astrocytes: signalling pathways, gene profiles and neuroprotective role in brain injury. Tesis doctoral Luis Pardo Fernández Bellaterra, Septiembre 2015 Instituto de Neurociencias Departamento de Bioquímica i Biologia Molecular Unidad de Bioquímica y Biologia Molecular Facultad de Medicina CREB-dependent transcription in astrocytes: signalling pathways, gene profiles and neuroprotective role in brain injury. Memoria del trabajo experimental para optar al grado de doctor, correspondiente al Programa de Doctorado en Neurociencias del Instituto de Neurociencias de la Universidad Autónoma de Barcelona, llevado a cabo por Luis Pardo Fernández bajo la dirección de la Dra. Elena Galea Rodríguez de Velasco y la Dra. Roser Masgrau Juanola, en el Instituto de Neurociencias de la Universidad Autónoma de Barcelona. Doctorando Directoras de tesis Luis Pardo Fernández Dra. Elena Galea Dra. Roser Masgrau In memoriam María Dolores Álvarez Durán Abuela, eres la culpable de que haya decidido recorrer el camino de la ciencia. Que estas líneas ayuden a conservar tu recuerdo. A mis padres y hermanos, A Meri INDEX I Summary 1 II Introduction 3 1 Astrocytes: physiology and pathology 5 1.1 Anatomical organization 6 1.2 Origins and heterogeneity 6 1.3 Astrocyte functions 8 1.3.1 Developmental functions 8 1.3.2 Neurovascular functions 9 1.3.3 Metabolic support 11 1.3.4 Homeostatic functions 13 1.3.5 Antioxidant functions 15 1.3.6 Signalling functions 15 1.4 Astrocytes in brain pathology 20 1.5 Reactive astrogliosis 22 2 The transcription -

Genetic Studies of Familial Vesicoureteral Reflux
Genetic Studies of Familial Vesicoureteral Reflux Zsuzsa Ingulf Bartik Department of Pediatrics Institute of Clinical Sciences Sahlgrenska Academy, University of Gothenburg Gothenburg 2018 Cover illustration: Searching for the VUR gene by Eszter Kónya Genetic Studies of Familial Vesicoureteral Reflux © Zsuzsa Ingulf Bartik 2018 [email protected] ISBN 978-91-7833-091-1 (PRINT) ISBN 978-91-7833-092-8 (PDF) Printed in Gothenburg, Sweden 2018 Printed by BrandFactory To my wonderful family Genetic Studies of Familial Vesicoureteral Reflux Zsuzsa Ingulf Bartik Department of Pediatrics, Institute of Clinical Sciences Sahlgrenska Academy, University of Gothenburg Gothenburg, Sweden ABSTRACT Vesicoureteral reflux (VUR) is a common congenital anomaly with a high risk of recurrent urinary tract infections (UTI) and, as a consequence, scarring of the renal parenchyma. Additionally, high-grade reflux is often associated with congenital renal damage (hypodysplasia). A clear heredity is seen, although genetic factors are only known for a minority of cases. The aim of this thesis was to study the heritability and genetic contribution as well as to compare the differences between familial and sporadic VUR. Study I compared clinical data from familial VUR with sporadic cases. Out of the 726 children with reflux that have been treated at Queen Silvia Children's Hospital between 1990 and 2004, 99 individuals (from 66 families) have reported relatives with VUR. A strong overrepresentation of maternal transmission of VUR was seen. The phenotype of VUR did not differ between familial and non-familial cases. Study II investigated the contribution of ROBO2 and SLIT2 genes in familial VUR through mutation screening by direct sequencing in 54 unrelated patients with primary VUR. -

Role of Maternal Sin3a in Reprogramming Gene Expression During Mouse Preimplantation Development
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2016 Role Of Maternal Sin3a In Reprogramming Gene Expression During Mouse Preimplantation Development Richard A. Jimenez University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Developmental Biology Commons, and the Genetics Commons Recommended Citation Jimenez, Richard A., "Role Of Maternal Sin3a In Reprogramming Gene Expression During Mouse Preimplantation Development" (2016). Publicly Accessible Penn Dissertations. 2366. https://repository.upenn.edu/edissertations/2366 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2366 For more information, please contact [email protected]. Role Of Maternal Sin3a In Reprogramming Gene Expression During Mouse Preimplantation Development Abstract In mouse, the maternal-to-zygotic transition entails a dramatic reprogramming of gene expression during the course of zygotic genome activation, which is essential for continued development beyond the 2-cell stage. Superimposed on zygotic genome activation and reprogramming of gene expression is formation of a chromatin-mediated transcriptionally repressive state that promotes repression of genes at the 2-cell stage. Experimentally inducing global histone hyperacetylation relieves this repression and histone deacetylase 1 (HDAC1) is the major HDAC involved in the development of this transcriptionally repressive state. Because SIN3A is essential -

Identification and Mechanistic Investigation of Recurrent Functional Genomic and Transcriptional Alterations in Advanced Prostat
Identification and Mechanistic Investigation of Recurrent Functional Genomic and Transcriptional Alterations in Advanced Prostate Cancer Thomas A. White A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2013 Reading Committee: Peter S. Nelson, Chair Janet L. Stanford Raymond J. Monnat Jay A. Shendure Program Authorized to Offer Degree: Molecular and Cellular Biology ©Copyright 2013 Thomas A. White University of Washington Abstract Identification and Mechanistic Investigation of Recurrent Functional Genomic and Transcriptional Alterations in Advanced Prostate Cancer Thomas A. White Chair of the Supervisory Committee: Peter S. Nelson, MD Novel functionally altered transcripts may be recurrent in prostate cancer (PCa) and may underlie lethal and advanced disease and the neuroendocrine small cell carcinoma (SCC) phenotype. We conducted an RNASeq survey of the LuCaP series of 24 PCa xenograft tumors from 19 men, and validated observations on metastatic tumors and PCa cell lines. Key findings include discovery and validation of 40 novel fusion transcripts including one recurrent chimera, many SCC-specific and castration resistance (CR) -specific novel splice isoforms, new observations on SCC-specific and TMPRSS2-ERG specific differential expression, the allele-specific expression of certain recurrent non- synonymous somatic single nucleotide variants (nsSNVs) previously discovered via exome sequencing of the same tumors, rgw ubiquitous A-to-I RNA editing of base excision repair (BER) gene NEIL1 as well as CDK13, a kinase involved in RNA splicing, and the SCC expression of a previously unannotated long noncoding RNA (lncRNA) at Chr6p22.2. Mechanistic investigation of the novel lncRNA indicates expression is regulated by derepression by master neuroendocrine regulator RE1-silencing transcription factor (REST), and may regulate some genes of axonogenesis and angiogenesis. -

De Novo Truncating Variants in PHF21A Cause Intellectual Disability and Craniofacial Anomalies
European Journal of Human Genetics (2019) 27:378–383 https://doi.org/10.1038/s41431-018-0289-x ARTICLE De novo truncating variants in PHF21A cause intellectual disability and craniofacial anomalies 1 2 3 4 5 Kohei Hamanaka ● Yuji Sugawara ● Takeyoshi Shimoji ● Tone Irene Nordtveit ● Mitsuhiro Kato ● 6 6 7 7 4 Mitsuko Nakashima ● Hirotomo Saitsu ● Toshimitsu Suzuki ● Kazuhiro Yamakawa ● Ingvild Aukrust ● 4 1 1 1 1 1 Gunnar Houge ● Satomi Mitsuhashi ● Atsushi Takata ● Kazuhiro Iwama ● Ahmed Alkanaq ● Atsushi Fujita ● 1 1 1 1,8 1 Eri Imagawa ● Takeshi Mizuguchi ● Noriko Miyake ● Satoko Miyatake ● Naomichi Matsumoto Received: 24 May 2018 / Revised: 26 September 2018 / Accepted: 28 September 2018 / Published online: 28 November 2018 © European Society of Human Genetics 2018 Abstract Potocki–Shaffer syndrome (PSS) is a contiguous gene syndrome caused by 11p11.2 deletions. PSS is clinically characterized by intellectual disability, craniofacial anomalies, enlarged parietal foramina, and multiple exostoses. PSS occasionally shows autism spectrum disorder, epilepsy, and overgrowth. Some of the clinical features are thought to be associated with haploinsufficiency of two genes in the 11p11.2 region; variants affecting the function of ALX4 cause enlarged parietal foramina and EXT2 lead to fi 1234567890();,: 1234567890();,: multiple exostoses. However, the remaining clinical features were still yet to be linked to speci c genetic alterations. In this study, we identified de novo truncating variants in an 11p11.2 gene, PHF21A, in three cases with intellectual disability and craniofacial anomalies. Among these three cases, autism spectrum disorder was recognized in one case, epilepsy in one case, and overgrowth in two cases. This study shows that PHF21A haploinsufficiency results in intellectual disability and craniofacial anomalies and possibly contributes to susceptibility to autism spectrum disorder, epilepsy, and overgrowth, all of which are PSS features. -

Aberrant DNA Methylation Profile Exacerbates Inflammation and Neurodegeneration in Multiple Sclerosis Patients Naiara Celarain* and Jordi Tomas-Roig*
Celarain and Tomas-Roig Journal of Neuroinflammation (2020) 17:21 https://doi.org/10.1186/s12974-019-1667-1 REVIEW Open Access Aberrant DNA methylation profile exacerbates inflammation and neurodegeneration in multiple sclerosis patients Naiara Celarain* and Jordi Tomas-Roig* Abstract Multiple sclerosis (MS) is an autoimmune and demyelinating disease of the central nervous system characterised by incoordination, sensory loss, weakness, changes in bladder capacity and bowel function, fatigue and cognitive impairment, creating a significant socioeconomic burden. The pathogenesis of MS involves both genetic susceptibility and exposure to distinct environmental risk factors. The gene x environment interaction is regulated by epigenetic mechanisms. Epigenetics refers to a complex system that modifies gene expression without altering the DNA sequence. The most studied epigenetic mechanism is DNA methylation. This epigenetic mark participates in distinct MS pathophysiological processes, including blood–brain barrier breakdown, inflammatory response, demyelination, remyelination failure and neurodegeneration. In this study, we also accurately summarised a list of environmental factors involved in the MS pathogenesis and its clinical course. A literature search was conducted using MEDLINE through PubMED and Scopus. In conclusion, an exhaustive study of DNA methylation might contribute towards new pharmacological interventions in MS by use of epigenetic drugs. Keywords: Multiple sclerosis, DNA methylation, Environmental risk factors, Inflammation, Neurodegeneration Background factors would be responsible for triggering autoimmun- Multiple sclerosis (MS) is an autoimmune, inflammatory, ity in MS patients. For this reason, the underlying mech- demyelinating and neurodegenerative disease of the cen- anisms involved in the MS pathogenesis can differ tral nervous system (CNS) [1]. As a result of myelin among patients. -

A Gene Implicated in Activation of Retinoic Acid Receptor Targets Is a Novel Renal Agenesis Gene in Humans
HIGHLIGHTED ARTICLE | INVESTIGATION A Gene Implicated in Activation of Retinoic Acid Receptor Targets Is a Novel Renal Agenesis Gene in Humans Patrick D. Brophy,*,1 Maria Rasmussen,†,1 Mrutyunjaya Parida,‡,1 Greg Bonde,§ Benjamin W. Darbro,* Xiaojing Hong,‡ Jason C. Clarke,* Kevin A. Peterson,** James Denegre,** Michael Schneider,†† Caroline R. Sussman,‡‡ Lone Sunde,† Dorte L. Lildballe,† Jens Michael Hertz,§§ Robert A. Cornell,§ Stephen A. Murray,** and J. Robert Manak*,‡,2 *Department of Pediatrics, ‡Department of Biology, and §Department of Anatomy and Cell Biology, University of Iowa, Iowa City, Iowa 52242, †Department of Clinical Genetics, Aarhus University Hospital, Skejby, Denmark 8200, **Genetics and Genomics, The Jackson Laboratory, Bar Harbor, Maine 04609, ††Medical Genetics, Carle Foundation Hospital and Physician Group, Urbana, Illinois 61801, ‡‡Department of Nephrology and Hypertension, Mayo Clinic, Rochester, Minnesota 55905, and §§Department of Clinical Genetics, Odense University Hospital, Odense, Denmark 5000 ABSTRACT Renal agenesis (RA) is one of the more extreme examples of congenital anomalies of the kidney and urinary tract (CAKUT). Bilateral renal agenesis is almost invariably fatal at birth, and unilateral renal agenesis can lead to future health issues including end- stage renal disease. Genetic investigations have identified several gene variants that cause RA, including EYA1, LHX1, and WT1. However, whereas compound null mutations of genes encoding a and g retinoic acid receptors (RARs) cause RA in mice, to date there have been no reports of variants in RAR genes causing RA in humans. In this study, we carried out whole exome sequence analysis of two families showing inheritance of an RA phenotype, and in both identified a single candidate gene, GREB1L. -

Genome-Wide Association Study for Carcass Traits in an Experimental Nelore Cattle Population
RESEARCH ARTICLE Genome-Wide Association Study for Carcass Traits in an Experimental Nelore Cattle Population Rafael Medeiros de Oliveira Silva1, Nedenia Bonvino Stafuzza1, Breno de Oliveira Fragomeni2, Grego rio Miguel Ferreira de Camargo1, ThaõÂs Matos Ceacero3, Joslaine Noely dos Santos GoncËalves Cyrillo3, Fernando Baldi1, Arione Augusti Boligon4, Maria Eugênia Zerlotti Mercadante3, Daniela Lino Lourenco2, Ignacy Misztal2, Lucia Galvão de Albuquerque1* a1111111111 1 School of Agricultural and Veterinarian Sciences, FCAV/ UNESP±Sao Paulo State University, Department of Animal Science, Jaboticabal-SP, Brazil, 2 University of Georgia, Department of Animal and Dairy Science, a1111111111 Athens, GA, United States of America, 3 APTA Center of Beef Cattle, Animal Science Institute, Sertaozinho, a1111111111 SP, Brazil, 4 Department of Animal Science, Federal University of Pelotas, Pelotas, RS, Brazil a1111111111 a1111111111 * [email protected] Abstract OPEN ACCESS The purpose of this study was to identify genomic regions associated with carcass traits in Citation: Medeiros de Oliveira Silva R, Bonvino an experimental Nelore cattle population. The studied data set contained 2,306 ultrasound Stafuzza N, de Oliveira Fragomeni B, Miguel records for longissimus muscle area (LMA), 1,832 for backfat thickness (BF), and 1,830 for Ferreira de Camargo G, Matos Ceacero T, Noely dos Santos GoncËalves Cyrillo J, et al. (2017) rump fat thickness (RF). A high-density SNP panel (BovineHD BeadChip assay 700k, Illu- Genome-Wide Association Study for Carcass Traits mina Inc., San Diego, CA) was used for genotyping. After genomic data quality control, in an Experimental Nelore Cattle Population. PLoS 437,197 SNPs from 761 animals were available, of which 721 had phenotypes for LMA, 669 ONE 12(1): e0169860. -
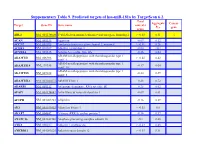
Suppementary Table 9. Predicted Targets of Hsa-Mir-181A by Targetscan 6.2
Suppementary Table 9. Predicted targets of hsa-miR-181a by TargetScan 6.2. Total Aggregate Cancer Target Gene ID Gene name context+ P gene score CT ABL2 NM_001136000 V-abl Abelson murine leukemia viral oncogene homolog 2 > -0.03 0.31 √ ACAN NM_001135 Aggrecan -0.09 0.12 ACCN2 NM_001095 Amiloride-sensitive cation channel 2, neuronal > -0.01 0.26 ACER3 NM_018367 Alkaline ceramidase 3 -0.04 <0.1 ACVR2A NM_001616 Activin A receptor, type IIA -0.16 0.64 ADAM metallopeptidase with thrombospondin type 1 ADAMTS1 NM_006988 > -0.02 0.42 motif, 1 ADAM metallopeptidase with thrombospondin type 1 ADAMTS18 NM_199355 -0.17 0.64 motif, 18 ADAM metallopeptidase with thrombospondin type 1 ADAMTS5 NM_007038 -0.24 0.59 motif, 5 ADAMTSL1 NM_001040272 ADAMTS-like 1 -0.26 0.72 ADARB1 NM_001112 Adenosine deaminase, RNA-specific, B1 -0.28 0.62 AFAP1 NM_001134647 Actin filament associated protein 1 -0.09 0.61 AFTPH NM_001002243 Aftiphilin -0.16 0.49 AK3 NM_001199852 Adenylate kinase 3 > -0.02 0.6 AKAP7 NM_004842 A kinase (PRKA) anchor protein 7 -0.16 0.37 ANAPC16 NM_001242546 Anaphase promoting complex subunit 16 -0.1 0.66 ANK1 NM_000037 Ankyrin 1, erythrocytic > -0.03 0.46 ANKRD12 NM_001083625 Ankyrin repeat domain 12 > -0.03 0.31 ANKRD33B NM_001164440 Ankyrin repeat domain 33B -0.17 0.35 ANKRD43 NM_175873 Ankyrin repeat domain 43 -0.16 0.65 ANKRD44 NM_001195144 Ankyrin repeat domain 44 -0.17 0.49 ANKRD52 NM_173595 Ankyrin repeat domain 52 > -0.05 0.7 AP1S3 NM_001039569 Adaptor-related protein complex 1, sigma 3 subunit -0.26 0.76 Amyloid beta (A4) precursor protein-binding, family A, APBA1 NM_001163 -0.13 0.81 member 1 APLP2 NM_001142276 Amyloid beta (A4) precursor-like protein 2 -0.05 0.55 APOO NM_024122 Apolipoprotein O -0.32 0.41 ARID2 NM_152641 AT rich interactive domain 2 (ARID, RFX-like) -0.07 0.55 √ ARL3 NM_004311 ADP-ribosylation factor-like 3 > -0.03 0.51 ARRDC3 NM_020801 Arrestin domain containing 3 > -0.02 0.47 ATF7 NM_001130059 Activating transcription factor 7 > -0.01 0.26 ATG2B NM_018036 ATG2 autophagy related 2 homolog B (S. -

New Insights Into Potocki-Shaffer Syndrome: Report of Two Novel
brain sciences Review New Insights into Potocki-Shaffer Syndrome: Report of Two Novel Cases and Literature Review Slavica Trajkova 1 , Eleonora Di Gregorio 2, Giovanni Battista Ferrero 3, Diana Carli 3 , Lisa Pavinato 1 , Geoffroy Delplancq 4,5 , Paul Kuentz 6,7,8 and Alfredo Brusco 1,2,* 1 Department of Medical Sciences, University of Torino, 10126 Turin, Italy; [email protected] (S.T.); [email protected] (L.P.) 2 Medical Genetics Unit, Città della Salute e della Scienza, University Hospital, 10126 Turin, Italy; [email protected] (E.D.) 3 Department of Public Health and Paediatrics, University of Torino, 10126 Turin, Italy; [email protected] (G.B.F.); [email protected] (D.C.) 4 Centre de Génétique Humaine, Université de Franche-Comté, 25000 Besançon, France; [email protected] (G.D.) 5 Service de Pédiatrie, CHU, 25000 Besançon, France 6 Oncobiologie Génétique Bioinformatique, PCBio, Centre Hospitalier Universitaire de Besançon, 25000 Besançon, France; [email protected] (P.K.) 7 UMR-Inserm 1231 GAD, Génétique des Anomalies du développement, Université de Bourgogne Franche-Comté, 21000 Dijon, France 8 Fédération Hospitalo-Universitaire Médecine Translationnelle et Anomalies du Développement (FHU TRANSLAD), Centre Hospitalier Universitaire de Dijon et Université de Bourgogne Franche-Comté, 21000 Dijon, France * Correspondence: [email protected] (A.B.) Received: 31 August 2020; Accepted: 27 October 2020; Published: 28 October 2020 Abstract: Potocki-Shaffer syndrome (PSS) is a rare non-recurrent contiguous gene deletion syndrome involving chromosome 11p11.2. Current literature implies a minimal region with haploinsufficiency of three genes, ALX4 (parietal foramina), EXT2 (multiple exostoses), and PHF21A (craniofacial anomalies, and intellectual disability).