Genome-Wide Association Study for Carcass Traits in an Experimental Nelore Cattle Population
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Data
Figure 2S 4 7 A - C 080125 CSCs 080418 CSCs - + IFN-a 48 h + IFN-a 48 h + IFN-a 72 h 6 + IFN-a 72 h 3 5 MRFI 4 2 3 2 1 1 0 0 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 7 B 13 080125 FBS - D 080418 FBS - + IFN-a 48 h 12 + IFN-a 48 h + IFN-a 72 h + IFN-a 72 h 6 080125 FBS 11 10 5 9 8 4 7 6 3 MRFI 5 4 2 3 2 1 1 0 0 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 MHC I MHC II MICA MICB ULBP-1 ULBP-2 ULBP-3 ULBP-4 Molecule Molecule FIGURE 4S FIGURE 5S Panel A Panel B FIGURE 6S A B C D Supplemental Results Table 1S. Modulation by IFN-α of APM in GBM CSC and FBS tumor cell lines. Molecule * Cell line IFN-α‡ HLA β2-m# HLA LMP TAP1 TAP2 class II A A HC§ 2 7 10 080125 CSCs - 1∞ (1) 3 (65) 2 (91) 1 (2) 6 (47) 2 (61) 1 (3) 1 (2) 1 (3) + 2 (81) 11 (80) 13 (99) 1 (3) 8 (88) 4 (91) 1 (2) 1 (3) 2 (68) 080125 FBS - 2 (81) 4 (63) 4 (83) 1 (3) 6 (80) 3 (67) 2 (86) 1 (3) 2 (75) + 2 (99) 14 (90) 7 (97) 5 (75) 7 (100) 6 (98) 2 (90) 1 (4) 3 (87) 080418 CSCs - 2 (51) 1 (1) 1 (3) 2 (47) 2 (83) 2 (54) 1 (4) 1 (2) 1 (3) + 2 (81) 3 (76) 5 (75) 2 (50) 2 (83) 3 (71) 1 (3) 2 (87) 1 (2) 080418 FBS - 1 (3) 3 (70) 2 (88) 1 (4) 3 (87) 2 (76) 1 (3) 1 (3) 1 (2) + 2 (78) 7 (98) 5 (99) 2 (94) 5 (100) 3 (100) 1 (4) 2 (100) 1 (2) 070104 CSCs - 1 (2) 1 (3) 1 (3) 2 (78) 1 (3) 1 (2) 1 (3) 1 (3) 1 (2) + 2 (98) 8 (100) 10 (88) 4 (89) 3 (98) 3 (94) 1 (4) 2 (86) 2 (79) * expression of APM molecules was evaluated by intracellular staining and cytofluorimetric analysis; ‡ cells were treatead or not (+/-) for 72 h with 1000 IU/ml of IFN-α; # β-2 microglobulin; § β-2 microglobulin-free HLA-A heavy chain; ∞ values are indicated as ratio between the mean of fluorescence intensity of cells stained with the selected mAb and that of the negative control; bold values indicate significant MRFI (≥ 2). -

Mutations in Diphosphoinositol- Pentakisphosphate Kinase PPIP5K2 Are Associated with Hearing Loss in Human and Mouse
RESEARCH ARTICLE Mutations in Diphosphoinositol- Pentakisphosphate Kinase PPIP5K2 are associated with hearing loss in human and mouse Rizwan Yousaf1, Chunfang Gu2, Zubair M. Ahmed3, Shaheen N. Khan4, Thomas B. Friedman5, Sheikh Riazuddin6, Stephen B. Shears2, Saima Riazuddin1* a1111111111 1 Laboratory of Molecular Genetics, Department of Otorhinolaryngology-Head & Neck Surgery, School of Medicine University of Maryland, Baltimore, MD, United States of America, 2 Signal Transduction Laboratory, a1111111111 National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, a1111111111 NC, United States of America, 3 Laboratory of Neurogenetics and Regenerative Medicine, Department of a1111111111 Otorhinolaryngology-Head & Neck Surgery, School of Medicine University of Maryland, Baltimore, MD, United a1111111111 States of America, 4 National Center for Excellence in Molecular Biology, University of the Punjab, Lahore, Pakistan, 5 Section on Human Genetics, Laboratory of Molecular Genetics, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Bethesda, MD, United States of America, 6 Shaheed Zulfiqar Ali Bhutto Medical University, Pakistan Institute of Medical Sciences, Islamabad, Pakistan * [email protected] OPEN ACCESS Citation: Yousaf R, Gu C, Ahmed ZM, Khan SN, Friedman TB, Riazuddin S, et al. (2018) Mutations Abstract in Diphosphoinositol-Pentakisphosphate Kinase PPIP5K2 are associated with hearing loss in Autosomal recessive nonsyndromic hearing loss is a genetically heterogeneous disorder. human and mouse. PLoS Genet 14(3): e1007297. https://doi.org/10.1371/journal.pgen.1007297 Here, we report a severe-to-profound sensorineural hearing loss locus, DFNB100 on chro- mosome 5q13.2-q23.2. Exome enrichment followed by massive parallel sequencing Editor: Karen B. Avraham, Tel Aviv University, ISRAEL revealed a c.2510G>A transition variant in PPIP5K2 that segregated with DFNB100-associ- ated hearing loss in two large apparently unrelated Pakistani families. -

Evidence for Differential Alternative Splicing in Blood of Young Boys With
Stamova et al. Molecular Autism 2013, 4:30 http://www.molecularautism.com/content/4/1/30 RESEARCH Open Access Evidence for differential alternative splicing in blood of young boys with autism spectrum disorders Boryana S Stamova1,2,5*, Yingfang Tian1,2,4, Christine W Nordahl1,3, Mark D Shen1,3, Sally Rogers1,3, David G Amaral1,3 and Frank R Sharp1,2 Abstract Background: Since RNA expression differences have been reported in autism spectrum disorder (ASD) for blood and brain, and differential alternative splicing (DAS) has been reported in ASD brains, we determined if there was DAS in blood mRNA of ASD subjects compared to typically developing (TD) controls, as well as in ASD subgroups related to cerebral volume. Methods: RNA from blood was processed on whole genome exon arrays for 2-4–year-old ASD and TD boys. An ANCOVA with age and batch as covariates was used to predict DAS for ALL ASD (n=30), ASD with normal total cerebral volumes (NTCV), and ASD with large total cerebral volumes (LTCV) compared to TD controls (n=20). Results: A total of 53 genes were predicted to have DAS for ALL ASD versus TD, 169 genes for ASD_NTCV versus TD, 1 gene for ASD_LTCV versus TD, and 27 genes for ASD_LTCV versus ASD_NTCV. These differences were significant at P <0.05 after false discovery rate corrections for multiple comparisons (FDR <5% false positives). A number of the genes predicted to have DAS in ASD are known to regulate DAS (SFPQ, SRPK1, SRSF11, SRSF2IP, FUS, LSM14A). In addition, a number of genes with predicted DAS are involved in pathways implicated in previous ASD studies, such as ROS monocyte/macrophage, Natural Killer Cell, mTOR, and NGF signaling. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

1 a Gene Implicated in Activation of Retinoic Acid Receptor Targets Is A
Genetics: Early Online, published on July 24, 2017 as 10.1534/genetics.117.1125 A gene implicated in activation of retinoic acid receptor targets is a novel renal agenesis gene in humans. Patrick D. Brophy, *1 Maria Rasmussen,†1 Mrutyunjaya Parida, ‡1 Greg Bonde, § Benjamin W. Darbro, * Xiaojing Hong, ‡ Jason C. Clarke, * Kevin A. Peterson, ** James Denegre, ** Michael Schneider, †† Caroline R. Sussman, ‡‡ Lone Sunde, † Dorte L. Lildballe, † Jens Michael Hertz, §§ Robert A. Cornell, §, Stephen A. Murray, ** J. Robert Manak*‡ 1Co-first authors *Department of Pediatrics, University of Iowa, Iowa City, IA, 52242. †Department of Clinical Genetics, Aarhus University Hospital, Skejby, Denmark ‡Department of Biology, University of Iowa, Iowa City, IA, 52242. §Department of Anatomy and Cell Biology, University of Iowa, IA, 52242. **The Jackson Laboratory, Bar Harbor, ME, 04609. ††Medical Genetics, Carle Foundation Hospital and Physician Group, Urbana, IL, 61801. ‡‡Department of Nephrology and Hypertension, Mayo Clinic, Rochester, MN, 55905. §§Department of Clinical Genetics, Odense University Hospital, Denmark IRB # 200711705 Whole exome sequencing data available in the Sequence Read Archive (SRA) database (accession number SRP112780) 1 Copyright 2017. Running Title: GREB1L, a novel renal agenesis gene Key Words: GREB1L, retinoic acid, renal agenesis, CAKUT, whole exome sequencing Corresponding author: J. Robert Manak, PhD 459 Biology Building, Dept. of Biology University of Iowa 129 E. Jefferson St. Iowa City, IA 52242 Ph. 319-335-0180 [email protected] 2 ABSTRACT Renal agenesis (RA) is one of the more extreme examples of congenital anomalies of the kidney and urinary tract (CAKUT). Bilateral renal agenesis is almost invariably fatal at birth, and unilateral renal agenesis can lead to future health issues including end stage renal disease. -

Análise Integrativa De Perfis Transcricionais De Pacientes Com
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Ribeirão Preto – 2012 ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Genética Orientador: Prof. Dr. Eduardo Antonio Donadi Co-orientador: Prof. Dr. Geraldo A. S. Passos Ribeirão Preto – 2012 AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE. FICHA CATALOGRÁFICA Evangelista, Adriane Feijó Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. Ribeirão Preto, 2012 192p. Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de Concentração: Genética. Orientador: Donadi, Eduardo Antonio Co-orientador: Passos, Geraldo A. 1. Expressão gênica – microarrays 2. Análise bioinformática por module maps 3. Diabetes mellitus tipo 1 4. Diabetes mellitus tipo 2 5. Diabetes mellitus gestacional FOLHA DE APROVAÇÃO ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. -

(A) Co-IP of HA Tagged ARMC9 with Flag Tagged Interactors
Supplemental Figures Supplemental Figure 1 Validation of ARMC9 interactome. (A) Co-IP of HA tagged ARMC9 with Flag tagged interactors. HA tagged ARMC9 was transfected alone or in conjunction with Flag tagged interactor constructs, HA beads were used to pull down the bait construct and blots were probed for the presence of the Flag tagged interactors. Experiment was performed in biological triplicates. Full blots are viewable in Plemental Figure 10. (B) Single transfection of PalmMyr-CFP-ARMC9 (green) and (C) single transfection of mRFP-TOGARAM1 (red) shows the localization in the absence of the respective interactor. (D) Co-expression of mRFP-TOGARAM1 and PalmMyr-CFP-ARMC9 shows microtubule colocalization. Scale bar indicates 20 μm. 1 Supplemental Figure 2 In silico modeling of the TOG2 variants. (A) Ribbon model of the wild type TOG2 domain structure of TOGARAM1 generated using HOPE. The protein is colored by the following elements: alpha-helices are show in blue, beta-strand is shown in red, turns are green, 3/10 helix is yellow, and random coil is cyan. (B) p. Arg368Trp missense variant in the TOG2 domain in ribbon-presentation generated using HOPE. TOG2 is shown in grey, the side chain of the mutated residue is shown in magenta. (C) A close up of the side chain of both wild type and mutated residue is shown in green and red respectively. (D) Inverted view of wild type TOG2 domain structure of TOGARAM1 as compared to (a). (E) p.Leu375Pro missense variant modeled in the TOG2 domain of TOGARAM1 in ribbon-presentation. TOG2 is shown in grey, the side chain of the mutated residue is shown in magenta. -

A CRISPR-Based Screen for Hedgehog Signaling Provides Insights Into Ciliary Function and Ciliopathies
ARTICLES https://doi.org/10.1038/s41588-018-0054-7 A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies David K. Breslow 1,2,7*, Sascha Hoogendoorn 3,7, Adam R. Kopp2, David W. Morgens4, Brandon K. Vu2, Margaret C. Kennedy1, Kyuho Han4, Amy Li4, Gaelen T. Hess4, Michael C. Bassik4, James K. Chen 3,5* and Maxence V. Nachury 2,6* Primary cilia organize Hedgehog signaling and shape embryonic development, and their dysregulation is the unifying cause of ciliopathies. We conducted a functional genomic screen for Hedgehog signaling by engineering antibiotic-based selection of Hedgehog-responsive cells and applying genome-wide CRISPR-mediated gene disruption. The screen can robustly identify factors required for ciliary signaling with few false positives or false negatives. Characterization of hit genes uncovered novel components of several ciliary structures, including a protein complex that contains δ -tubulin and ε -tubulin and is required for centriole maintenance. The screen also provides an unbiased tool for classifying ciliopathies and showed that many congenital heart disorders are caused by loss of ciliary signaling. Collectively, our study enables a systematic analysis of ciliary function and of ciliopathies, and also defines a versatile platform for dissecting signaling pathways through CRISPR-based screening. he primary cilium is a surface-exposed microtubule-based approach. Indeed, most studies to date have searched for genes that compartment that serves as an organizing center for diverse either intrinsically affect cell growth or affect sensitivity to applied Tsignaling pathways1–3. Mutations affecting cilia cause ciliopa- perturbations16–23. thies, a group of developmental disorders including Joubert syn- Here, we engineered a Hh-pathway-sensitive reporter to enable drome, Meckel syndrome (MKS), nephronophthisis (NPHP), and an antibiotic-based selection platform. -

Identification of Potential Key Genes and Pathway Linked with Sporadic Creutzfeldt-Jakob Disease Based on Integrated Bioinformatics Analyses
medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of potential key genes and pathway linked with sporadic Creutzfeldt-Jakob disease based on integrated bioinformatics analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract Sporadic Creutzfeldt-Jakob disease (sCJD) is neurodegenerative disease also called prion disease linked with poor prognosis. The aim of the current study was to illuminate the underlying molecular mechanisms of sCJD. The mRNA microarray dataset GSE124571 was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened. -

Prioritization of Metabolic Genes As Novel Therapeutic Targets in Estrogen-Receptor Negative Breast Tumors Using Multi-Omics Data and Text Mining
www.oncotarget.com Oncotarget, 2019, Vol. 10, (No. 39), pp: 3894-3909 Research Paper Prioritization of metabolic genes as novel therapeutic targets in estrogen-receptor negative breast tumors using multi-omics data and text mining Dinesh Kumar Barupal1,*, Bei Gao1,*, Jan Budczies2, Brett S. Phinney4, Bertrand Perroud4, Carsten Denkert2,3 and Oliver Fiehn1 1West Coast Metabolomics Center, University of California, Davis, CA, USA 2Institute of Pathology, Charité University Hospital, Berlin, Germany 3German Institute of Pathology, Philipps-University Marburg, Marburg, Germany 4UC Davis Genome Center, University of California, Davis, CA, USA *Co-first authors and contributed equally to this work Correspondence to: Oliver Fiehn, email: [email protected] Keywords: set-enrichment; ChemRICH; multi-omics; metabolic networks; candidate gene prioritization Received: March 12, 2019 Accepted: May 13, 2019 Published: June 11, 2019 Copyright: Barupal et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Estrogen-receptor negative (ERneg) breast cancer is an aggressive breast cancer subtype in the need for new therapeutic options. We have analyzed metabolomics, proteomics and transcriptomics data for a cohort of 276 breast tumors (MetaCancer study) and nine public transcriptomics datasets using univariate statistics, meta- analysis, Reactome pathway analysis, biochemical network mapping and text mining of metabolic genes. In the MetaCancer cohort, a total of 29% metabolites, 21% proteins and 33% transcripts were significantly different (raw p <0.05) between ERneg and ERpos breast tumors. -
Next-MP50 Status Report
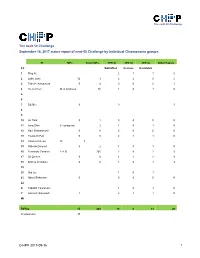
The neXt-50 Challenge The neXt-50 Challenge September 16, 2017 status report of next-50 Challenge by individual Chromosome groups. PI MPs Silver MPs JPR SI JPR SI JPR SI Other Papers Ch Submitted In press In revision 1 Ping Xu 2 1 1 0 2 Lydie Lane 12 3 2 2 0 2 3 Takeshi Kawamura 0 4 0 0 0 1 4 Yu-Ju Chen 26 in progress 86 1 0 1 0 5 6 7 Ed Nice 0 0 1 8 9 10 Jin Park 0 1 0 0 0 0 11 Jong Shin 2 in progress 2 1 0 1 0 12 Ravi Sirdeshmukh 0 0 0 0 0 0 13 Young-Ki Paik 0 5 2 1 1 0 14 CharLes Pineau 12 3 15 GiLberto Domont 0 2 1 0 1 0 16 Fernando CorraLes 2 (+ 9) 165 2 0 2 5 17 GiL Omenn 0 0 3 1 2 8 18 Andrey Archakov 0 0 1 0 1 3 19 20 Siqi Liu 1 0 1 21 Albert Sickmann 0 0 0 0 0 22 X Tadashi Yamamoto 1 0 1 0 Y Hosseini SaLekdeh 1 2 1 1 0 Mt TOTAL 15 268 19 6 13 20 (in progress) 37 C-HPP 2017-09-16 1 The neXt-50 Challenge Chromosome 1 (Ping Xu) PIC Leaders: Ping Xu, Fuchu He Contributing labs: Ping Xu, Beijing Proteome Research Center Fuchu He, Beijing Proteome Research Center Dong Yang, Beijing Proteome Research Center Wantao Ying, Beijing Proteome Research Center Pengyuan Yang, Fudan University Siqi Liu, Beijing Genome Institute Qinyu He, Jinan University Major lab members or partners contributing to the neXt50: Yao Zhang (Beijing Proteome Research Center), Yihao Wang (Beijing Proteome Research Center), Cuitong He (Beijing Proteome Research Center), Wei Wei (Beijing Proteome Research Center), Yanchang Li (Beijing Proteome Research Center), Feng Xu (Beijing Proteome Research Center), Xuehui Peng (Beijing Proteome Research Center). -

Limitations of the Rhesus Macaque Draft Genome Assembly and Annotation
University of Nebraska Medical Center DigitalCommons@UNMC Journal Articles: Genetics, Cell Biology & Anatomy Genetics, Cell Biology & Anatomy 2012 Limitations of the Rhesus Macaque Draft Genome Assembly and Annotation Xiongfei Zhang University of Nebraska Medical Center Joel Goodsell University of Nebraska Medical Center, [email protected] Robert B. Norgren University of Nebraska Medical Center, [email protected] Follow this and additional works at: https://digitalcommons.unmc.edu/com_gcba_articles Part of the Medical Anatomy Commons, Medical Cell Biology Commons, and the Medical Genetics Commons Recommended Citation Zhang, Xiongfei; Goodsell, Joel; and Norgren, Robert B., "Limitations of the Rhesus Macaque Draft Genome Assembly and Annotation" (2012). Journal Articles: Genetics, Cell Biology & Anatomy. 42. https://digitalcommons.unmc.edu/com_gcba_articles/42 This Article is brought to you for free and open access by the Genetics, Cell Biology & Anatomy at DigitalCommons@UNMC. It has been accepted for inclusion in Journal Articles: Genetics, Cell Biology & Anatomy by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected]. Zhang et al. BMC Genomics 2012, 13 :206 http://www.biomedcentral.com/1471-2164/13/206 CORRESPONDENCE Open Access Limitations of the rhesus macaque draft genome assembly and annotation Xiongfei Zhang, Joel Goodsell and Robert B Norgren, Jr. * Abstract Finished genome sequences and assemblies are available for only a few vertebrates. Thus, investigators studying many species must rely on draft genomes. Using the rhesus macaque as an example, we document the effects of sequencing errors, gaps in sequence and misassemblies on one automated gene model pipeline, Gnomon. The combination of draft genome with automated gene finding software can result in spurious sequences.