Transcriptome Analysis Identifies Novel Prognostic Genes in Osteosarcoma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Characterization of Dysregulated Lncrna-Mrna Network Based on Cerna Hypothesis to Reveal the Occurrence and Recurrence of Myocar
Zhang et al. Cell Death Discovery (2018) 4:35 DOI 10.1038/s41420-018-0036-7 Cell Death Discovery ARTICLE Open Access Characterization of dysregulated lncRNA- mRNA network based on ceRNA hypothesis to reveal the occurrence and recurrence of myocardial infarction Guangde Zhang1,HaoranSun2, Yawei Zhang2, Hengqiang Zhao2, Wenjing Fan1,JianfeiLi3,YingliLv2, Qiong Song2, Jiayao Li2,MingyuZhang1 and Hongbo Shi2 Abstract Accumulating evidence has demonstrated that long non-coding RNAs (lncRNAs) acting as competing endogenous RNAs (ceRNAs) play important roles in initiation and development of human diseases. However, the mechanism of ceRNA regulated by lncRNA in myocardial infarction (MI) remained unclear. In this study, we performed a multi-step computational method to construct dysregulated lncRNA-mRNA networks for MI occurrence (DLMN_MI_OC) and recurrence (DLMN_MI_Re) based on “ceRNA hypothesis”. We systematically integrated lncRNA and mRNA expression profiles and miRNA-target regulatory interactions. The constructed DLMN_MI_OC and DLMN_MI_Re both exhibited biological network characteristics, and functional analysis demonstrated that the networks were specific for MI. Additionally, we identified some lncRNA-mRNA ceRNA modules involved in MI occurrence and recurrence. Finally, two new panel biomarkers defined by four lncRNAs (RP1-239B22.5, AC135048.13, RP11-4O1.2, RP11-285F7.2) from 1234567890():,; 1234567890():,; DLMN_MI_OC and three lncRNAs (RP11-363E7.4, CTA-29F11.1, RP5-894A10.6) from DLMN_MI_Re with high classification performance were, respectively, identified in distinguishing controls from patients, and patients with recurrent events from those without recurrent events. This study will provide us new insight into ceRNA-mediated regulatory mechanisms involved in MI occurrence and recurrence, and facilitate the discovery of candidate diagnostic and prognosis biomarkers for MI. -

Germline Mutations Causing Familial Lung Cancer
Journal of Human Genetics (2015) 60, 597–603 & 2015 The Japan Society of Human Genetics All rights reserved 1434-5161/15 www.nature.com/jhg ORIGINAL ARTICLE Germline mutations causing familial lung cancer Koichi Tomoshige1,2, Keitaro Matsumoto1, Tomoshi Tsuchiya1, Masahiro Oikawa1, Takuro Miyazaki1, Naoya Yamasaki1, Hiroyuki Mishima2, Akira Kinoshita2, Toru Kubo3, Kiyoyasu Fukushima3, Koh-ichiro Yoshiura2 and Takeshi Nagayasu1 Genetic factors are important in lung cancer, but as most lung cancers are sporadic, little is known about inherited genetic factors. We identified a three-generation family with suspected autosomal dominant inherited lung cancer susceptibility. Sixteen individuals in the family had lung cancer. To identify the gene(s) that cause lung cancer in this pedigree, we extracted DNA from the peripheral blood of three individuals and from the blood of one cancer-free control family member and performed whole-exome sequencing. We identified 41 alterations in 40 genes in all affected family members but not in the unaffected member. These were considered candidate mutations for familial lung cancer. Next, to identify somatic mutations and/or inherited alterations in these 40 genes among sporadic lung cancers, we performed exon target enrichment sequencing using 192 samples from sporadic lung cancer patients. We detected somatic ‘candidate’ mutations in multiple sporadic lung cancer samples; MAST1, CENPE, CACNB2 and LCT were the most promising candidate genes. In addition, the MAST1 gene was located in a putative cancer-linked locus in the pedigree. Our data suggest that several genes act as oncogenic drivers in this family, and that MAST1 is most likely to cause lung cancer. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Single-Cell Transcriptome Profiling of the Kidney Glomerulus Identifies Key Cell Types and Reactions to Injury
BASIC RESEARCH www.jasn.org Single-Cell Transcriptome Profiling of the Kidney Glomerulus Identifies Key Cell Types and Reactions to Injury Jun-Jae Chung ,1 Leonard Goldstein ,2 Ying-Jiun J. Chen,2 Jiyeon Lee ,1 Joshua D. Webster,3 Merone Roose-Girma,2 Sharad C. Paudyal,4 Zora Modrusan,2 Anwesha Dey,5 and Andrey S. Shaw1 Due to the number of contributing authors, the affiliations are listed at the end of this article. ABSTRACT Background The glomerulus is a specialized capillary bed that is involved in urine production and BP control. Glomerular injury is a major cause of CKD, which is epidemic and without therapeutic options. Single-cell transcriptomics has radically improved our ability to characterize complex organs, such as the kidney. Cells of the glomerulus, however, have been largely underrepresented in previous single-cell kidney studies due to their paucity and intractability. Methods Single-cell RNA sequencing comprehensively characterized the types of cells in the glomerulus from healthy mice and from four different disease models (nephrotoxic serum nephritis, diabetes, doxo- rubicin toxicity, and CD2AP deficiency). Results Allcelltypesintheglomeruluswereidentified using unsupervised clustering analysis. Novel marker genes and gene signatures of mesangial cells, vascular smooth muscle cells of the afferent and efferent arteri- oles, parietal epithelial cells, and three types of endothelial cells were identified. Analysis of the disease models revealed cell type–specific and injury type–specific responses in the glomerulus, including acute activation of the Hippo pathway in podocytes after nephrotoxic immune injury. Conditional deletion of YAP or TAZ resulted in more severe and prolonged proteinuria in response to injury, as well as worse glomerulosclerosis. -

Genome-Wide DNA Methylation Dynamics During Epigenetic
Gómez‑Redondo et al. Clin Epigenet (2021) 13:27 https://doi.org/10.1186/s13148‑021‑01003‑x RESEARCH Open Access Genome‑wide DNA methylation dynamics during epigenetic reprogramming in the porcine germline Isabel Gómez‑Redondo1*† , Benjamín Planells1†, Sebastián Cánovas2,3, Elena Ivanova4, Gavin Kelsey4,5 and Alfonso Gutiérrez‑Adán1 Abstract Background: Prior work in mice has shown that some retrotransposed elements remain substantially methylated during DNA methylation reprogramming of germ cells. In the pig, however, information about this process is scarce. The present study was designed to examine the methylation profles of porcine germ cells during the time course of epigenetic reprogramming. Results: Sows were artifcially inseminated, and their fetuses were collected 28, 32, 36, 39, and 42 days later. At each time point, genital ridges were dissected from the mesonephros and germ cells were isolated through magnetic‑ activated cell sorting using an anti‑SSEA‑1 antibody, and recovered germ cells were subjected to whole‑genome bisulphite sequencing. Methylation levels were quantifed using SeqMonk software by performing an unbiased analysis, and persistently methylated regions (PMRs) in each sex were determined to extract those regions showing 50% or more methylation. Most genomic elements underwent a dramatic loss of methylation from day 28 to day 36, when the lowest levels were shown. By day 42, there was evidence for the initiation of genomic re‑methylation. We identifed a total of 1456 and 1122 PMRs in male and female germ cells, respectively, and large numbers of transpos‑ able elements (SINEs, LINEs, and LTRs) were found to be located within these PMRs. Twenty‑one percent of the introns located in these PMRs were found to be the frst introns of a gene, suggesting their regulatory role in the expression of these genes. -

Supplemental Data
SUPPLEMENTAL DATA A transcriptomic continuum of differentiation arrest identifies myeloid interface acute leukemias with poor prognosis Bond et al Contents: • Supplementary Table Legends • Supplementary Tables S6, S8 - S11 • Supplementary Figures S1 - S4 • Supplementary Methods • Supplementary References Supplementary Table Legends: Supplementary Table S1: Gene-sets used for GSEA analysis in this study (see Excel file). Supplementary Table S2: Details of patient cohort (see Excel file). Supplementary Table S3: Differential gene expression analysis comparing AML-like T- ALLs with other T-ALLs (see Excel file). Positive values denote genes with higher expression in AML-like T-ALLs. Supplementary Table S4: Differential gene expression analysis for the comparisons of thymic subset populations indicated in each tab (see Excel files). Supplementary Table S5: ICGS output (see Excel file). First row contains sample names, second row the ICGS clusters, and the following rows guide genes and their normalized expression. The second column indicates the guide genes groups as indicated by the black and white bars in Figure 2A (first tab), Supplementary Figure S3A (second tab (1)) and S3C (third tab (2)). Supplementary Table S6: Genes included in the targeted NGS panel. Supplementary Table S7: Mutational status by NGS (see Excel file). 0 = no mutation, 1 = mutation of known significance, 2 = mutation of unknown significance. Supplementary Table S8: List of genes used for IAL score Supplementary Table S9: Impact of IAL score on outcome according to ELN subgroup. Supplementary Table S10: Comparison of clinicobiological characteristics and mutational profiles of cases with high and low IAL scores in the ALFA-0701 cohort (3). Supplemental Table S11: Univariate analyses of Overall Survival in the ALFA-0701 cohort (3). -

A Single Genetic Locus Associated with Pediatric Fractures: a Genome-Wide Association Study on 3,230 Patients
1716 EXPERIMENTAL AND THERAPEUTIC MEDICINE 20: 1716-1724, 2020 A single genetic locus associated with pediatric fractures: A genome-wide association study on 3,230 patients ROOPE PARVIAINEN1, SINI SKARP2,3, LINDA KORHONEN1, WILLY SERLO1, MINNA MÄNNIKKÖ2 ��� JUHA-JAAKKO SINIKUMPU1 1D�p���m��� �� C������� ��� A����������, O��� C�������� F������� ��� Sp���� I�j��y S���y, R������� U��� ��� P���������, P�������� N�������y, P�������� Surgery, C���� P�y������y, D��m������y, C������� G�������, O��������� ��� Gy�������y, O����������y������y ��� Op�����m����y (PEDEGO), O��� M������ R������� C����� (MRC), U��������y �� O��� ��� O��� U��������y H��p����, FI-90029 O���; 2N������� F������ B���� C�����, F�����y �� M�������, U��������y �� O���; 3C����� ��� L��� C����� H����� R�������, F�����y �� M�������, U��������y �� O���, FI-90014 O���, F������ Received July 13, 2019; Accepted April 29, 2020 DOI: 10.3892/etm.2020.8885 Abstract. The�� understanding������������� ��of the��� biological���������� and��� environ�������- RNA 1 (PROSER2-AS1) and PROSER2, thus suggesting that mental risk factors of fractures in pediatrics is limited. Previous these m�y be novel candidate genes associated with the risk of studies have reported that fractures involve heritable traits, but pediatric fractures. the genetic factors contributing to the risk of fractures remain elusive. Furthermore, genetic influences specific to immature Introduction bone have not been thoroughly studied. Therefore, the aim of the present study was to identify genetic variations that are The incidence of fractures is highest �mong the young and associated with fractures in early childhood. The present study older p�pulations (1). O� note, fractures in pediatric patients used � prospective Northern Finland Birth Cohort (year 1986; are common injuries that result in pain, as well as short‑ and �=9,432). -

Cis-Regulatory Differences Explaining Evolved Levels of Endometrial Invasibility in Eutherian Mammals
bioRxiv preprint doi: https://doi.org/10.1101/2020.09.04.283366; this version posted September 5, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Cis-Regulatory Differences Explaining Evolved Levels of Endometrial Invasibility in Eutherian Mammals Yasir Suhail1,3, Jamie D. Maziarz2,3, Anasuya Dighe 2,3 , Gunter Wagner2,3,5,6,*, Kshitiz1,3,4,* 1 Department of Biomedical Engineering, University of Connecticut Health, Farmington, CT 2. Department of Ecology and Evolutionary Biology, Yale University, New Haven, CT 3. Cancer Systems Biology Center (CaSB@Yale), Yale University, West Haven, CT 4. Center for Cell Analysis and Modeling, University of Connecticut Health, Farmington, CT 4. Department of Obstetrics, Gynecology and Reproductive Sciences, Yale Medical School, New Haven, CT 5. Department of Obstetrics and Gynecology, Wayne State University, Detroit, MI * Corresponding Authors: [email protected], [email protected] Abstract Eutherian (placental) mammals exhibit great differences in the degree of placental invasion into the maternal endometrium, with humans being on the most invasive end. Previously, we have shown that these differences in invasiveness is largely controlled by the stromal fibroblasts of the maternal endometrium, with secondary effect on stroma of other tissues resulting in correlated differences in cancer malignancy. Here, we present a statistical investigation of the second dogma linking the phenotypic and transcriptional differences to the genomic changes across species, revealing the regulatory genomic sequence differences underlying these inter-species differences. -

Using Whole-Exome Sequencing and Protein Interaction Networks To
www.nature.com/scientificreports OPEN Using whole‑exome sequencing and protein interaction networks to prioritize candidate genes for germline cutaneous melanoma susceptibility Sally Yepes1*, Margaret A. Tucker1, Hela Koka1, Yanzi Xiao1, Kristine Jones1,2, Aurelie Vogt1,2, Laurie Burdette1,2, Wen Luo1,2, Bin Zhu1,2, Amy Hutchinson1,2, Meredith Yeager1,2, Belynda Hicks1,2, Neal D. Freedman1, Stephen J. Chanock1, Alisa M. Goldstein1,3 & Xiaohong R. Yang1,3* Although next‑generation sequencing has demonstrated great potential for novel gene discovery, confrming disease‑causing genes after initial discovery remains challenging. Here, we applied a network analysis approach to prioritize candidate genes identifed from whole‑exome sequencing analysis of 98 cutaneous melanoma patients from 27 families. Using a network propagation method, we ranked candidate genes by their similarity to known disease genes in protein–protein interaction networks and identifed gene clusters with functional connectivity. Using this approach, we identifed several new candidate susceptibility genes that warrant future investigations such as NGLY1, IL1RN, FABP2, PRKDC, and PROSER2. The propagated network analysis also allowed us to link families that did not have common underlying genes but that carried variants in genes that interact on protein– protein interaction networks. In conclusion, our study provided an analysis perspective for gene prioritization in the context of genetic heterogeneity across families and prioritized top potential candidate susceptibility genes in our dataset. CDKN2A and CDK4 are the two well-established high-risk genes for familial cutaneous malignant melanoma (CMM). In the last several years, BAP1, POT1, ACD, TERF2IP, and TERT were also identifed as high-risk melanoma susceptibility genes1. -

Microrna-31 Negatively Regulates Peripherally Derived Regulatory T-Cell Generation by Repressing Retinoic Acid-Inducible Protein 3
ARTICLE Received 17 Feb 2015 | Accepted 27 May 2015 | Published 13 Jul 2015 DOI: 10.1038/ncomms8639 OPEN MicroRNA-31 negatively regulates peripherally derived regulatory T-cell generation by repressing retinoic acid-inducible protein 3 Lingyun Zhang1, Fang Ke1, Zhaoyuan Liu1, Jing Bai1, Jinlin Liu1, Sha Yan1, Zhenyao Xu1, Fangzhou Lou1, Hong Wang1, Huiyuan Zhu1, Yang Sun1, Wei Cai1, Yuanyuan Gao1, Qun Li2, Xue-Zhong Yu3, Youcun Qian4, Zichun Hua5, Jiong Deng6, Qi-Jing Li7 & Honglin Wang1,8 Peripherally derived regulatory T (pTreg) cell generation requires T-cell receptor (TCR) signalling and the cytokines TGF-b1 and IL-2. Here we show that TCR signalling induces the microRNA miR-31, which negatively regulates pTreg-cell generation. miR-31 conditional deletion results in enhanced induction of pTreg cells, and decreased severity of experimental autoimmune encephalomyelitis (EAE). Unexpectedly, we identify Gprc5a as a direct target of miR-31. Gprc5a is known as retinoic acid-inducible protein 3, and its deficiency leads to impaired pTreg-cell induction and increased EAE severity. By generating miR-31 and Gprc5a double knockout mice, we show that miR-31 promotes the development of EAE through inhibiting Gprc5a. Thus, our data identify miR-31 and its target Gprc5a as critical regulators for pTreg-cell generation, suggesting a previously unrecognized epigenetic mechanism for dysfunctional Treg cells in autoimmune diseases. 1 Shanghai Institute of Immunology, Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education, Shanghai Jiao Tong University School of Medicine (SJTU-SM), Shanghai 200025, China. 2 Shanghai Institute of Hypertension, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine (SJTU-SM), Shanghai 200025, China. -

SUPPLEMENTARY DATA Retinoic Acid Mediates Visceral-Specific
SUPPLEMENTARY DATA Retinoic Acid Mediates Visceral-specific Adipogenic Defects of Human Adipose-derived Stem Cells Kosuke Takeda, Sandhya Sriram, Xin Hui Derryn Chan, Wee Kiat Ong, Chia Rou Yeo, Betty Tan, Seung-Ah Lee, Kien Voon Kong, Shawn Hoon, Hongfeng Jiang, Jason J. Yuen, Jayakumar Perumal, Madhur Agrawal, Candida Vaz, Jimmy So, Asim Shabbir, William S. Blaner, Malini Olivo, Weiping Han, Vivek Tanavde, Sue-Anne Toh, and Shigeki Sugii Supplementary Experimental Procedures RNA-Seq experiment and analysis Total RNA was extracted from SC and VS fat depots using Qiagen RNeasy plus kit. Poly-A mRNA was then enriched from ~5 µg of total RNA with oligo dT beads (Life Technologies). Approximately 100 ng of poly-A mRNA recovered was used to construct multiplexed strand-specific RNA-seq libraries as per manufacturer’s instruction (NEXTflexTM Rapid Directional RNA-SEQ Kit (dUTP-Based) v2). Individual library quality was assessed with an Agilent 2100 Bioanalyzer and quantified with a QuBit 2.0 fluorometer before pooling for sequencing on Illumina HiSeq 2000 (1x101 bp read). The pooled libraries were quantified using the KAPA quantification kit (KAPA Biosystems) prior to cluster formation. Fastq formatted reads were processed with Trimmomatic to remove adapter sequences and trim low quality bases (LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36). Reads were aligned to the human genome (hg19) using Tophat version 2 (settings --no-coverage-search --library- type=fr-firststrand). Feature read counts were generated using htseq-count (Python package HTSeq default union-counting mode, strand=reverse). Differential Expression analysis was performed using the edgeR package in both ‘classic’ and generalized linear model (glm) modes to contrast SC and VS adipose tissues from 8 non-diabetic patients. -
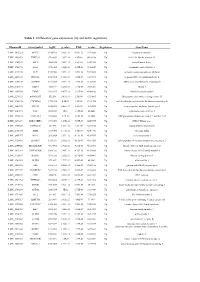
Table 1. Differential Gene Expression (Up and Down Regulation)
Table 1. Differential gene expression (up and down regulation). IlluminaId GeneSymbol logFC p value FDR t value Regulation GeneName ILMN_1814221 NPTX1 8120.166 2.02E-16 9.56E-12 107.0626 Up neuronal pentraxin 1 ILMN_1693471 KLHL35 879.4869 2.03E-14 4.8E-10 66.98158 Up kelch like family member 35 ILMN_1705153 NEFH 3866.938 3.55E-14 5.6E-10 63.27518 Up neurofilament heavy ILMN_1708778 ASS1 3751.105 6.81E-14 6.69E-10 59.20647 Up argininosuccinate synthase 1 ILMN_2114720 SLPI 1949.583 9.25E-14 7.29E-10 57.39296 Up secretory leukocyte peptidase inhibitor ILMN_2415583 HS6ST2 150.4964 1.51E-13 1.02E-09 54.59292 Up heparansulfate 6-O-sulfotransferase 2 ILMN_3185198 ACTR3C 112.0705 2.58E-13 1.53E-09 51.68897 Up ARP3 actin-related protein 3 homolog C ILMN_1700541 FBLN1 3126.79 3.36E-13 1.77E-09 50.31287 Up fibulin 1 ILMN_2109708 TYMP 922.6535 4.87E-13 2.31E-09 48.44836 Up thymidine phosphorylase ILMN_2214473 ARHGEF35 555.516 5.87E-13 2.5E-09 47.53665 Up Rho guanine nucleotide exchange factor 35 ILMN_1740170 CHCHD10 1770.965 6.4E-13 2.5E-09 47.11754 Up coiled-coil-helix-coiled-coil-helix domain containing 10 ILMN_1800739 SPINT2 1656.074 6.86E-13 2.5E-09 46.78296 Up serine peptidase inhibitor, Kunitz type 2 ILMN_3241522 ASS1 603.9339 1E-12 3.15E-09 45.0058 Up argininosuccinate synthase 1 ILMN_1740233 UGT1A10 540.0026 1.1E-12 3.15E-09 44.5868 Up UDP glucuronosyltransferase family 1 member A10 ILMN_3273229 MIR193BHG 273.3476 1.13E-12 3.15E-09 44.45755 Up MIR193B host gene ILMN_1748827 TMEM238 614.575 1.65E-12 3.8E-09 42.78856 Up transmembrane protein