BMC Evolutionary Biology Biomed Central
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Competing Generic Concepts for Blanding's, Pacific and European
Zootaxa 2791: 41–53 (2011) ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ Article ZOOTAXA Copyright © 2011 · Magnolia Press ISSN 1175-5334 (online edition) Competing generic concepts for Blanding’s, Pacific and European pond turtles (Emydoidea, Actinemys and Emys)—Which is best? UWE FRITZ1,3, CHRISTIAN SCHMIDT1 & CARL H. ERNST2 1Museum of Zoology, Senckenberg Dresden, A. B. Meyer Building, D-01109 Dresden, Germany 2Division of Amphibians and Reptiles, MRC 162, Smithsonian Institution, P.O. Box 37012, Washington, D.C. 20013-7012, USA 3Corresponding author. E-mail: [email protected] Abstract We review competing taxonomic classifications and hypotheses for the phylogeny of emydine turtles. The formerly rec- ognized genus Clemmys sensu lato clearly is paraphyletic. Two of its former species, now Glyptemys insculpta and G. muhlenbergii, constitute a well-supported basal clade within the Emydinae. However, the phylogenetic position of the oth- er two species traditionally placed in Clemmys remains controversial. Mitochondrial data suggest a clade embracing Actinemys (formerly Clemmys) marmorata, Emydoidea and Emys and as its sister either another clade (Clemmys guttata + Terrapene) or Terrapene alone. In contrast, nuclear genomic data yield conflicting results, depending on which genes are used. Either Clemmys guttata is revealed as sister to ((Emydoidea + Emys) + Actinemys) + Terrapene or Clemmys gut- tata is sister to Actinemys marmorata and these two species together are the sister group of (Emydoidea + Emys); Terra- pene appears then as sister to (Actinemys marmorata + Clemmys guttata) + (Emydoidea + Emys). The contradictory branching patterns depending from the selected loci are suggestive of lineage sorting problems. Ignoring the unclear phy- logenetic position of Actinemys marmorata, one recently proposed classification scheme placed Actinemys marmorata, Emydoidea blandingii, Emys orbicularis, and Emys trinacris in one genus (Emys), while another classification scheme treats Actinemys, Emydoidea, and Emys as distinct genera. -

Redalyc.MORPHOLOGY and CONSERVATION of the MESOAMERICAN SLIDER (Trachemys Venusta, Emydidae) from the ATRATO RIVER BASIN, COLOMB
Acta Biológica Colombiana ISSN: 0120-548X [email protected] Universidad Nacional de Colombia Sede Bogotá Colombia CEBALLOS, CLAUDIA P.; BRAND, WILLIAM A. MORPHOLOGY AND CONSERVATION OF THE MESOAMERICAN SLIDER (Trachemys venusta, Emydidae) FROM THE ATRATO RIVER BASIN, COLOMBIA Acta Biológica Colombiana, vol. 19, núm. 3, septiembre-diciembre, 2014, pp. 483-488 Universidad Nacional de Colombia Sede Bogotá Bogotá, Colombia Available in: http://www.redalyc.org/articulo.oa?id=319031647014 How to cite Complete issue Scientific Information System More information about this article Network of Scientific Journals from Latin America, the Caribbean, Spain and Portugal Journal's homepage in redalyc.org Non-profit academic project, developed under the open access initiative SEDE BOGOTÁ ACTA BIOLÓGICA COLOMBIANA FACULTAD DE CIENCIAS DEPARTAMENTO DE BIOLOGÍA ARTÍCULO DE INVESTIGACIÓN MORPHOLOGY AND CONSERVATION OF THE MESOAMERICAN SLIDER (Trachemys venusta, EMYDIDAE) FROM THE ATRATO RIVER BASIN, COLOMBIA Morfología y conservación de la tortuga hicotea Mesoamericana (Trachemys venusta, Emydidae) del río Atrato, Colombia CLAUDIA P. CEBALLOS1, Ph. D.; WILLIAM A. BRAND2, Ecol. 1 Grupo Centauro. Escuela de Medicina Veterinaria, Facultad de Ciencias Agrarias, Universidad de Antioquia. Carrera 75 n.º 65-87, of. 47- 122, Medellín, Colombia. [email protected] 2 Corpouraba. Calle 92 n.º 98-39, Turbo, Antioquia, Colombia. [email protected] Author for correspondence: Claudia P. Ceballos, [email protected] Received 20th February 2014, first decision 14th May 2014, accepted 05th June 2014. Citation / Citar este artículo como: CEBALLOS CP, BRAND WA. Morphology and conservation of the mesoamerican slider (Trachemys venusta, Emydidae) from the Atrato River basin, Colombia. Acta biol. Colomb. 2014;19(3):483-488 ABSTRACT The phylogenetic relationships of the Mesoamerican Slider, Trachemys venusta, that inhabits the Atrato River basin of Colombia have been controversial as three different names have been proposed during the last 12 years: T. -

Xenosaurus Tzacualtipantecus. the Zacualtipán Knob-Scaled Lizard Is Endemic to the Sierra Madre Oriental of Eastern Mexico
Xenosaurus tzacualtipantecus. The Zacualtipán knob-scaled lizard is endemic to the Sierra Madre Oriental of eastern Mexico. This medium-large lizard (female holotype measures 188 mm in total length) is known only from the vicinity of the type locality in eastern Hidalgo, at an elevation of 1,900 m in pine-oak forest, and a nearby locality at 2,000 m in northern Veracruz (Woolrich- Piña and Smith 2012). Xenosaurus tzacualtipantecus is thought to belong to the northern clade of the genus, which also contains X. newmanorum and X. platyceps (Bhullar 2011). As with its congeners, X. tzacualtipantecus is an inhabitant of crevices in limestone rocks. This species consumes beetles and lepidopteran larvae and gives birth to living young. The habitat of this lizard in the vicinity of the type locality is being deforested, and people in nearby towns have created an open garbage dump in this area. We determined its EVS as 17, in the middle of the high vulnerability category (see text for explanation), and its status by the IUCN and SEMAR- NAT presently are undetermined. This newly described endemic species is one of nine known species in the monogeneric family Xenosauridae, which is endemic to northern Mesoamerica (Mexico from Tamaulipas to Chiapas and into the montane portions of Alta Verapaz, Guatemala). All but one of these nine species is endemic to Mexico. Photo by Christian Berriozabal-Islas. amphibian-reptile-conservation.org 01 June 2013 | Volume 7 | Number 1 | e61 Copyright: © 2013 Wilson et al. This is an open-access article distributed under the terms of the Creative Com- mons Attribution–NonCommercial–NoDerivs 3.0 Unported License, which permits unrestricted use for non-com- Amphibian & Reptile Conservation 7(1): 1–47. -
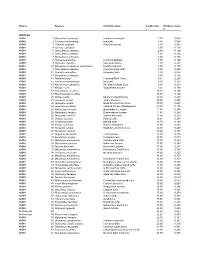
PDF File Containing Table of Lengths and Thicknesses of Turtle Shells And
Source Species Common name length (cm) thickness (cm) L t TURTLES AMNH 1 Sternotherus odoratus common musk turtle 2.30 0.089 AMNH 2 Clemmys muhlenbergi bug turtle 3.80 0.069 AMNH 3 Chersina angulata Angulate tortoise 3.90 0.050 AMNH 4 Testudo carbonera 6.97 0.130 AMNH 5 Sternotherus oderatus 6.99 0.160 AMNH 6 Sternotherus oderatus 7.00 0.165 AMNH 7 Sternotherus oderatus 7.00 0.165 AMNH 8 Homopus areolatus Common padloper 7.95 0.100 AMNH 9 Homopus signatus Speckled tortoise 7.98 0.231 AMNH 10 Kinosternon subrabum steinochneri Florida mud turtle 8.90 0.178 AMNH 11 Sternotherus oderatus Common musk turtle 8.98 0.290 AMNH 12 Chelydra serpentina Snapping turtle 8.98 0.076 AMNH 13 Sternotherus oderatus 9.00 0.168 AMNH 14 Hardella thurgi Crowned River Turtle 9.04 0.263 AMNH 15 Clemmys muhlenbergii Bog turtle 9.09 0.231 AMNH 16 Kinosternon subrubrum The Eastern Mud Turtle 9.10 0.253 AMNH 17 Kinixys crosa hinged-back tortoise 9.34 0.160 AMNH 18 Peamobates oculifers 10.17 0.140 AMNH 19 Peammobates oculifera 10.27 0.140 AMNH 20 Kinixys spekii Speke's hinged tortoise 10.30 0.201 AMNH 21 Terrapene ornata ornate box turtle 10.30 0.406 AMNH 22 Terrapene ornata North American box turtle 10.76 0.257 AMNH 23 Geochelone radiata radiated tortoise (Madagascar) 10.80 0.155 AMNH 24 Malaclemys terrapin diamondback terrapin 11.40 0.295 AMNH 25 Malaclemys terrapin Diamondback terrapin 11.58 0.264 AMNH 26 Terrapene carolina eastern box turtle 11.80 0.259 AMNH 27 Chrysemys picta Painted turtle 12.21 0.267 AMNH 28 Chrysemys picta painted turtle 12.70 0.168 AMNH 29 -

The PARI Journal Vol. VI, No. 3
ThePARIJournal A quarterly publication of the Pre-Columbian Art Research Institute Volume VI, No.3, Winter 2006 Teasing the Turtle from its Shell: In This Issue: AHKand MAHK in MayaWriting* Teasing the Turtle MARC ZENDER from its Shell: Peabody Museum of Archaeology & Ethnology, Harvard University AHK and MAHK in Maya Writing resents the profile carapace of a testudine by Marc Zender (i.e., a turtle, tortoise or terrapin), careful PAGES 1-14 examination of their formal features, dis- tributions and lexical associations reveals • them to have been quite distinct entities, Morley's Diary: rigidly distinguished by scribes through- a b out the life of the script. As discussed in April 23, 1932 detail below, there is in fact strong reason Figure 1. “Turtle Shell” Glyphs: (a) MAHK “carapace” and PAGES 15-16 “cover”; (b) AHK “turtle.” All drawings by the author. to see the first sign (Figure 1a) as the de- piction of a turtle shell carrying the value Formally similar but nevertheless dis- MAHK “carapace, shell,” and, less specifi- tinct signs remain a constant challenge to cally, “cover, enclosure.” By contrast, the Joel Skidmore students of the ancient Maya script. Pre- latter sign (Figure 1b), while also depict- Editor ing a turtle shell, has long been known [email protected] viously I have argued that a number of signs traditionally considered identical to carry the value AHK “turtle,” and is are in fact quite distinct, as demonstrated therefore best thought of as the pars pro The PARI Journal by formal divergences and consistent dis- toto depiction of a “turtle” proper. -

Terrapene Carolina (Linnaeus 1758) – Eastern Box Turtle, Common Box Turtle
Conservation Biology of Freshwater Turtles and Tortoises: A Compilation Project ofEmydidae the IUCN/SSC — TortoiseTerrapene and Freshwatercarolina Turtle Specialist Group 085.1 A.G.J. Rhodin, P.C.H. Pritchard, P.P. van Dijk, R.A. Saumure, K.A. Buhlmann, J.B. Iverson, and R.A. Mittermeier, Eds. Chelonian Research Monographs (ISSN 1088-7105) No. 5, doi:10.3854/crm.5.085.carolina.v1.2015 © 2015 by Chelonian Research Foundation • Published 26 January 2015 Terrapene carolina (Linnaeus 1758) – Eastern Box Turtle, Common Box Turtle A. ROSS KIESTER1 AND LISABETH L. WILLEY2 1Turtle Conservancy, 49 Bleecker St., Suite 601, New York, New York 10012 USA [[email protected]]; 2Department of Environmental Studies, Antioch University New England, 40 Avon St., Keene, New Hampshire 03431 USA [[email protected]] SUMMARY. – The Eastern Box Turtle, Terrapene carolina (Family Emydidae), as currently understood, contains six living subspecies of small turtles (carapace lengths to ca. 115–235 mm) able to close their hinged plastrons into a tightly closed box. Although the nominate subspecies is among the most widely distributed and well-known of the world’s turtles, the two Mexican subspecies are poorly known. This primarily terrestrial, though occasionally semi-terrestrial, species ranges throughout the eastern and southern United States and disjunctly in Mexico. It was generally recognized as common in the USA throughout the 20th century, but is now threatened by continuing habitat conversion, road mortality, and collection for the pet trade, and notable population declines have been documented throughout its range. In the United States, this turtle is a paradigm example of the conservation threats that beset and impact a historically common North American species. -

Aspects of the Life History of the Texas Map Turtle (Graptemys Versa)
Am. Midl. Nat. 153:378–388 Aspects of the Life History of the Texas Map Turtle (Graptemys versa) PETER V. LINDEMAN1 Department of Biology and Health Services, 150 Cooper Hall, Edinboro University of Pennsylvania, Edinboro 16444 ABSTRACT.—The Texas map turtle (Graptemys versa) is endemic to the Colorado River drainage in southcentral Texas. A study of its life history was undertaken using data collected in 1998–2000 from a population in the South Llano River, southernmost tributary of the Colorado drainage, and data from museum specimens that had been collected from the South Llano River in 1949. Compared to congeners, G. versa is a small-bodied species. Its small body size is, predictably, linked to relatively small clutch size, small egg size, rapid growth toward asymptotic size and early maturation. As many as four clutches may be laid during an active season, although the effects of follicular atresia on clutch frequency are not known. Both clutch size and egg width were positively correlated with female body size, with the former relationship having a log-log slope significantly less than the expected value of 3, probably due to the latter relationship. Analyses were consistent with the hypothesis of anatomical constraint on egg size, with at least smaller females laying eggs that are of less than optimal size. No differences were found in body size or clutch size between 1949 and 1998–2000 despite a large- scale change in diet associated with invasion of the river by Asian clams (Corbicula sp.). However, body size is substantially reduced in the South Llano River compared to other sections of the Colorado drainage, a finding mimicked by at least one other turtle species in the drainage, Pseudemys texana. -

Abstract Keywords First Records of 5 Allochthonous Species and Subspecies of Turtles (Trachemys Scripta Troostii , Mauremys Casp
ISSN 1989-8649 Manag. Biolog. Invasions, 2011, 2 Abstract First records of 5 allochthonous species and subspecies of Turtles The aim of the research was to (Trachemys scripta troostii, Mauremys caspica, Mauremys rivulata, investigate the presence of Pelodiscus sinensis, Testudo horsfieldii) and new records of subspecies allochthonous species of turtles in the Trachemys scripta elegans in Latvia nature of Latvia, as well as to withdraw Mihails PUPINS & Aija PUPINA them from the nature according to the "Plan on protection of Emys orbicularis in Latvia", officially approved by the Introduction, Hypotheses and appearance of allochthonous Latvian Ministry of Environment (Pupiņš Problems for Management species of turtles in the nature of & Pupiņa 2007a). The methods of the Latvia (Pupiņš & Pupiņa 2007b). research were: 1) information campaign Anthropogenic spreading and and survey; 2) interviews with invasion of allochthonous (Kabish Hypotheses of the research. inhabitants; 3) field expeditions; 4) 1990) species of reptiles is an issue The main hypothesis of the research laboratory study of viability of found of world importance for preserving that has been carried out since 2004 turtles. In course of research, five new natural biodiversity (Kraus 2009). is based on the assumption that allochthonous species and subspecies of Allochthonous species of reptile there are also allochthonous turtles the turtles were recorded for the first reproduce in new habitats (Cadi et in the nature of Latvia besides time, and new data were obtained autochthonous turtles. The about finding Trachemys scripta elegans al. 2004; Heeswijk-Dihter et al. in the nature of Latvia. Most of the 2006; Perez-Santigosa et al. 2008) following assumptions have been registered animals (80%) were removed competing with ecologically close formulated as additional from the nature. -

A Systematic Review of the Turtle Family Emydidae
67 (1): 1 – 122 © Senckenberg Gesellschaft für Naturforschung, 2017. 30.6.2017 A Systematic Review of the Turtle Family Emydidae Michael E. Seidel1 & Carl H. Ernst 2 1 4430 Richmond Park Drive East, Jacksonville, FL, 32224, USA and Department of Biological Sciences, Marshall University, Huntington, WV, USA; [email protected] — 2 Division of Amphibians and Reptiles, mrc 162, Smithsonian Institution, P.O. Box 37012, Washington, D.C. 200137012, USA; [email protected] Accepted 19.ix.2016. Published online at www.senckenberg.de / vertebrate-zoology on 27.vi.2016. Abstract Family Emydidae is a large and diverse group of turtles comprised of 50 – 60 extant species. After a long history of taxonomic revision, the family is presently recognized as a monophyletic group defined by unique skeletal and molecular character states. Emydids are believed to have originated in the Eocene, 42 – 56 million years ago. They are mostly native to North America, but one genus, Trachemys, occurs in South America and a second, Emys, ranges over parts of Europe, western Asia, and northern Africa. Some of the species are threatened and their future survival depends in part on understanding their systematic relationships and habitat requirements. The present treatise provides a synthesis and update of studies which define diversity and classification of the Emydidae. A review of family nomenclature indicates that RAFINESQUE, 1815 should be credited for the family name Emydidae. Early taxonomic studies of these turtles were based primarily on morphological data, including some fossil material. More recent work has relied heavily on phylogenetic analyses using molecular data, mostly DNA. The bulk of current evidence supports two major lineages: the subfamily Emydinae which has mostly semi-terrestrial forms ( genera Actinemys, Clemmys, Emydoidea, Emys, Glyptemys, Terrapene) and the more aquatic subfamily Deirochelyinae ( genera Chrysemys, Deirochelys, Graptemys, Malaclemys, Pseudemys, Trachemys). -

Investigations of the Ecology of Graptemys Versa and Pseudemys
LINNAEUS FUND RESEARCH REPORTS 223 aged 5.6 eggs (range 4-9 eggs). Average egg dimensions for Che Ionian Conservation and Biology , 200 I, 4( I ):223-224 Cl 200 I by Chclonian Research Foundation 32 eggs obtained by oxytocin injection were 35.26 mm for length(range33.0-38.4mm)and20.87mmforwidth(range Investigation s of the Ecology of 18.4-22.2 mm). Graptemys versa and Pseudemys texana Mature male P. texana ranged from 78-162 mm PL in Southcentral Texas. (mean 122.2 mm, n =62). Female P. texana ranged from 95- Linnaeus Fund Research Report 234 mm PL (mean 178.8 mm, n = 54). The minimum size of gravid females was 213 mm PL and the mean size of females 1 this size or larger was 222.6 mm PL (n = 13). Clutch size of PETER V. LINDEMAN four females radio graphed in 1999 averaged 8.3 eggs (range 1Department of Biology and Health Services, 7-9eggs). Average egg dimensions for23 eggs obtained by 150 Cooper Hall, Edinboro University of Pennsylvania, oxytocin injection were 42.19 mm for length (range 39.9- Edinboro, Pennsylvania, 16444 USA 44.7 mm) and 26.96 mm for width (range 26.0-27.6 mm). [Fax: 814-732-2593; E-mail: [email protected]] Diet.- Mollusks were abundant in the diet of 12 female G. versa from 1949 and 16 females captured in 1998- 99. Ernst et al. (1994) published an exhaustive compen Insects were abundant in the diet of male G. versa (which are dium of literature citations for the chelonian fauna of North much smaller than females, as in other Graptemys; Ernst et al., America. -

Studies on the Molecular Underpinnings of Sex Determination Mechanism Evolution and Molecular Sexing Tools in Turtles Robert Alan Literman Iowa State University
Iowa State University Capstones, Theses and Graduate Theses and Dissertations Dissertations 2017 Studies on the molecular underpinnings of sex determination mechanism evolution and molecular sexing tools in turtles Robert Alan Literman Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/etd Part of the Developmental Biology Commons, Evolution Commons, and the Molecular Biology Commons Recommended Citation Literman, Robert Alan, "Studies on the molecular underpinnings of sex determination mechanism evolution and molecular sexing tools in turtles" (2017). Graduate Theses and Dissertations. 15350. https://lib.dr.iastate.edu/etd/15350 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Studies on the molecular underpinnings of sex determination mechanism evolution and molecular sexing tools in turtles by Robert Literman A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Ecology and Evolutionary Biology Program of Study Committee: Maria N Valenzuela-Castro, Major Professor Jeff Essner John Nason Michael Shogren-Knaak Jonathan Wendel Iowa State University Ames, Iowa 2017 Copyright © Robert Literman, 2017. All rights reserved. ii DEDICATION This work is dedicated to my family: To my parents who supported me in all endeavors leading here, and to my wife Krystal and our son Rubin, who kept me going through it all. -

Chelonian Advisory Group Regional Collection Plan 4Th Edition December 2015
Association of Zoos and Aquariums (AZA) Chelonian Advisory Group Regional Collection Plan 4th Edition December 2015 Editor Chelonian TAG Steering Committee 1 TABLE OF CONTENTS Introduction Mission ...................................................................................................................................... 3 Steering Committee Structure ........................................................................................................... 3 Officers, Steering Committee Members, and Advisors ..................................................................... 4 Taxonomic Scope ............................................................................................................................. 6 Space Analysis Space .......................................................................................................................................... 6 Survey ........................................................................................................................................ 6 Current and Potential Holding Table Results ............................................................................. 8 Species Selection Process Process ..................................................................................................................................... 11 Decision Tree ........................................................................................................................... 13 Decision Tree Results .............................................................................................................