Comparative Analysis of Rodent and Small Mammal Viromes to Better
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Identifing Priority Ecoregions for Rodent Conservation at the Genus Level
Oryx Vol 35 No 2 April 2001 Short Communication Identifing priority ecoregions for rodent conservation at the genus level Giovanni Amori and Spartaco Gippoliti Abstract Rodents account for 40 per cent of living high number of genera) 'threat-spots' for rodent conser- mammal species. Nevertheless, despite an increased vation. A few regions, mainly drylands, are singled out interest in biodiversity conservation and their high as important areas for rodent conservation but are not species richness, Rodentia are often neglected by con- generally recognized in global biodiversity assessments. servationists. We attempt for the first time a world-wide These are the remaining forests of Togo, extreme evaluation of rodent conservation priorities at the genus 'western Sahel', the Turanian and Mongolian-Manchu- level. Given the low popularity of the order, we rian steppes and the desert of the Horn of Africa. considered it desirable to discuss identified priorities Resources for conservation must be allocated first to within the framework of established biodiversity prior- recognized threat spots and to those restricted-range ity areas of the world. Two families and 62 genera are genera which may depend on species-specific strategies recognized as threatened. Our analyses highlight the for their survival. Philippines, New Guinea, Sulawesi, the Caribbean, China temperate forests and the Atlantic Forest of Keywords Biodiversity, conservation priorities, south-eastern Brazil as the most important (for their rodents, threatened genera, world ecoregions. Conservation efforts for rodents must be included in Introduction the general framework of mammalian diversity conser- With 26-32 recognized extant families and more than vation, focusing on a biodiversity/area approach. -

Genome Sequencing by Random Priming Methods for Viral Identification
Genome sequencing by random priming methods for viral identification Rosseel Toon Dissertation submitted in fulfillment of the requirements for the degree of Doctor of Philosophy (PhD) in Veterinary Sciences, Faculty of Veterinary Medicine, Ghent University, 2015 Promotors: Dr. Steven Van Borm Prof. Dr. Hans Nauwynck “The real voyage of discovery consist not in seeking new landscapes, but in having new eyes” Marcel Proust, French writer, 1923 Table of contents Table of contents ....................................................................................................................... 1 List of abbreviations ................................................................................................................. 3 Chapter 1 General introduction ................................................................................................ 5 1. Viral diagnostics and genomics ....................................................................................... 7 2. The DNA sequencing revolution ................................................................................... 12 2.1. Classical Sanger sequencing .................................................................................. 12 2.2. Next-generation sequencing ................................................................................... 16 3. The viral metagenomic workflow ................................................................................. 24 3.1. Sample preparation ............................................................................................... -

Redalyc.Mountain Vizcacha (Lagidium Cf. Peruanum) in Ecuador
Mastozoología Neotropical ISSN: 0327-9383 [email protected] Sociedad Argentina para el Estudio de los Mamíferos Argentina Werner, Florian A.; Ledesma, Karim J.; Hidalgo B., Rodrigo Mountain vizcacha (Lagidium cf. peruanum) in Ecuador - First record of chinchillidae from the northern Andes Mastozoología Neotropical, vol. 13, núm. 2, julio-diciembre, 2006, pp. 271-274 Sociedad Argentina para el Estudio de los Mamíferos Tucumán, Argentina Available in: http://www.redalyc.org/articulo.oa?id=45713213 How to cite Complete issue Scientific Information System More information about this article Network of Scientific Journals from Latin America, the Caribbean, Spain and Portugal Journal's homepage in redalyc.org Non-profit academic project, developed under the open access initiative Mastozoología Neotropical, 13(2):271-274, Mendoza, 2006 ISSN 0327-9383 ©SAREM, 2006 Versión on-line ISSN 1666-0536 www.cricyt.edu.ar/mn.htm MOUNTAIN VIZCACHA (LAGIDIUM CF. PERUANUM) IN ECUADOR – FIRST RECORD OF CHINCHILLIDAE FROM THE NORTHERN ANDES Florian A. Werner¹, Karim J. Ledesma2, and Rodrigo Hidalgo B.3 1 Albrecht-von-Haller-Institute of Plant Sciences, University of Göttingen, Untere Karspüle 2, 37073 Göttingen, Germany; <[email protected]>. 2 Department of Biological Sciences, Florida Atlantic University, Boca Raton, U.S.A; <[email protected]>. 3 Colegio Nacional Eloy Alfaro, Gonzales Suarez y Sucre, Cariamanga, Ecuador; <[email protected]>. Key words. Biogeography. Caviomorpha. Distribution. Hystricomorpha. Viscacha. Chinchillidae is a family of hystricomorph Cerro Ahuaca is a granite inselberg 2 km rodents distributed in the Andes of Peru, from the town of Cariamanga (1950 m), Loja Bolivia, Chile and Argentina, and in lowland province (4°18’29.4’’ S, 79°32’47.2’’ W). -

Evidence for Viral Infection in the Copepods Labidocera Aestiva And
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School January 2012 Evidence for Viral Infection in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida Darren Stephenson Dunlap University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the American Studies Commons, Other Oceanography and Atmospheric Sciences and Meteorology Commons, and the Virology Commons Scholar Commons Citation Dunlap, Darren Stephenson, "Evidence for Viral Infection in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida" (2012). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/4032 This Thesis is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Evidence of Viruses in the Copepods Labidocera aestiva and Acartia tonsa in Tampa Bay, Florida By Darren S. Dunlap A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science College of Marine Science University of South Florida Major Professor: Mya Breitbart, Ph.D Kendra Daly, Ph.D Ian Hewson, Ph.D Date of Approval: March 19, 2012 Key Words: Copepods, Single-stranded DNA Viruses, Mesozooplankton, Transmission Electron Microscopy, Metagenomics Copyright © 2012, Darren Stephenson Dunlap DEDICATION None of this would have been possible without the generous love and support of my entire family over the years. My parents, Steve and Jill Dunlap, have always encouraged my pursuits with support and love, and their persistence of throwing me into lakes and rivers is largely responsible for my passion for Marine Science. -

Guide for Common Viral Diseases of Animals in Louisiana
Sampling and Testing Guide for Common Viral Diseases of Animals in Louisiana Please click on the species of interest: Cattle Deer and Small Ruminants The Louisiana Animal Swine Disease Diagnostic Horses Laboratory Dogs A service unit of the LSU School of Veterinary Medicine Adapted from Murphy, F.A., et al, Veterinary Virology, 3rd ed. Cats Academic Press, 1999. Compiled by Rob Poston Multi-species: Rabiesvirus DCN LADDL Guide for Common Viral Diseases v. B2 1 Cattle Please click on the principle system involvement Generalized viral diseases Respiratory viral diseases Enteric viral diseases Reproductive/neonatal viral diseases Viral infections affecting the skin Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. B2 2 Deer and Small Ruminants Please click on the principle system involvement Generalized viral disease Respiratory viral disease Enteric viral diseases Reproductive/neonatal viral diseases Viral infections affecting the skin Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. B2 3 Swine Please click on the principle system involvement Generalized viral diseases Respiratory viral diseases Enteric viral diseases Reproductive/neonatal viral diseases Viral infections affecting the skin Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. B2 4 Horses Please click on the principle system involvement Generalized viral diseases Neurological viral diseases Respiratory viral diseases Enteric viral diseases Abortifacient/neonatal viral diseases Viral infections affecting the skin Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. B2 5 Dogs Please click on the principle system involvement Generalized viral diseases Respiratory viral diseases Enteric viral diseases Reproductive/neonatal viral diseases Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. -

Nucleotide Amino Acid Size (Nt) #Orfs Marnavirus Heterosigma Akashiwo Heterosigma Akashiwo RNA Heterosigma Lang Et Al
Supplementary Table 1: Summary of information for all viruses falling within the seven Marnaviridae genera in our analyses. Accession Genome Genus Species Virus name Strain Abbreviation Source Country Reference Nucleotide Amino acid Size (nt) #ORFs Marnavirus Heterosigma akashiwo Heterosigma akashiwo RNA Heterosigma Lang et al. , 2004; HaRNAV AY337486 AAP97137 8587 One Canada RNA virus 1 virus akashiwo Tai et al. , 2003 Marine single- ASG92540 Moniruzzaman et Classification pending Q sR OV 020 KY286100 9290 Two celled USA ASG92541 al ., 2017 eukaryotes Marine single- Moniruzzaman et Classification pending Q sR OV 041 KY286101 ASG92542 9328 One celled USA al ., 2017 eukaryotes APG78557 Classification pending Wenzhou picorna-like virus 13 WZSBei69459 KX884360 9458 One Bivalve China Shi et al ., 2016 APG78557 Classification pending Changjiang picorna-like virus 2 CJLX30436 KX884547 APG79001 7171 One Crayfish China Shi et al ., 2016 Beihai picorna-like virus 57 BHHQ57630 KX883356 APG76773 8518 One Tunicate China Shi et al ., 2016 Classification pending Beihai picorna-like virus 57 BHJP51916 KX883380 APG76812 8518 One Tunicate China Shi et al ., 2016 Marine single- ASG92530 Moniruzzaman et Classification pending N OV 137 KY130494 7746 Two celled USA ASG92531 al ., 2017 eukaryotes Hubei picorna-like virus 7 WHSF7327 KX884284 APG78434 9614 One Pill worm China Shi et al ., 2016 Classification pending Hubei picorna-like virus 7 WHCC111241 KX884268 APG78407 7945 One Insect China Shi et al ., 2016 Sanxia atyid shrimp virus 2 WHCCII13331 KX884278 APG78424 10445 One Insect China Shi et al ., 2016 Classification pending Freshwater atyid Sanxia atyid shrimp virus 2 SXXX37884 KX883708 APG77465 10400 One China Shi et al ., 2016 shrimp Labyrnavirus Aurantiochytrium single Aurantiochytrium single stranded BAE47143 Aurantiochytriu AuRNAV AB193726 9035 Three4 Japan Takao et al. -

Hantavirus Disease Were HPS Is More Common in Late Spring and Early Summer in Seropositive in One Study in the U.K
Hantavirus Importance Hantaviruses are a large group of viruses that circulate asymptomatically in Disease rodents, insectivores and bats, but sometimes cause illnesses in humans. Some of these agents can occur in laboratory rodents or pet rats. Clinical cases in humans vary in Hantavirus Fever, severity: some hantaviruses tend to cause mild disease, typically with complete recovery; others frequently cause serious illnesses with case fatality rates of 30% or Hemorrhagic Fever with Renal higher. Hantavirus infections in people are fairly common in parts of Asia, Europe and Syndrome (HFRS), Nephropathia South America, but they seem to be less frequent in North America. Hantaviruses may Epidemica (NE), Hantavirus occasionally infect animals other than their usual hosts; however, there is currently no Pulmonary Syndrome (HPS), evidence that they cause any illnesses in these animals, with the possible exception of Hantavirus Cardiopulmonary nonhuman primates. Syndrome, Hemorrhagic Nephrosonephritis, Epidemic Etiology Hemorrhagic Fever, Korean Hantaviruses are members of the genus Orthohantavirus in the family Hantaviridae Hemorrhagic Fever and order Bunyavirales. As of 2017, 41 species of hantaviruses had officially accepted names, but there is ongoing debate about which viruses should be considered discrete species, and additional viruses have been discovered but not yet classified. Different Last Updated: September 2018 viruses tend to be associated with the two major clinical syndromes in humans, hemorrhagic fever with renal syndrome (HFRS) and hantavirus pulmonary (or cardiopulmonary) syndrome (HPS). However, this distinction is not absolute: viruses that are usually associated with HFRS have been infrequently linked to HPS and vice versa. A mild form of HFRS in Europe is commonly called nephropathia epidemica. -

Enteric Viruses Nucleic Acids Distribution Along the Digestive Tract of Rhesus Macaques with Idiopathic Chronic Diarrhea
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.24.449827; this version posted June 24, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Enteric viruses nucleic acids distribution along the digestive tract of rhesus macaques with idiopathic chronic diarrhea Eric Delwart1,2*, David Merriam3,4, Amir Ardeshir3, Eda Altan1,2, Yanpeng Li1,2, Xutao Deng,1,2, J. Dennis Hartigan-O’Connor3 1. Vitlant Research Institute, 270 Masonic Ave, San Francisco CA94118 2. Dept of Laboratory Medicine, UCSF, San Francisco CA94118 3. California National Primate Research Center, University of California, Davis, CA 95616 4. Department of Pediatric Infectious Diseases, University of Colorado School of Medicine, Aurora, CO, USA. * Communicating author: [email protected] Abstract: Idiopathic chronic diarrhea (ICD) is a common clinical condition in captive rhesus macaques, claiming 33% of medical culls (i.e. deaths unrelated to research). Using viral metagenomics we characterized the eukaryotic virome in digestive tract tissues collected at necropsy from nine animals with ICD. We show the presence of multiple viruses in the Parvoviridae and Picornaviridae family. We then compared the distribution of viral reads in the stomach, duodenum, jejunum, ileum, and the proximal, transverse, and distal colons. Tissues and mucosal scraping from the same locations showed closely related results while different gut tissues from the same animal varied widely. Picornavirus reads were generally more abundant in the lower digestive tract, particularly in the descending (distal) colon. -

Porcine Kobuvirus
PORCINE KOBUVIRUS Prepared for the Swine Health Information Center By the Center for Food Security and Public Health, College of Veterinary Medicine, Iowa State University September 2015 SUMMARY Etiology • Porcine kobuvirus (PKoV) is a small, non-enveloped RNA virus in the family Picornaviridae. • There are three distinct clusters within the genus Kobuvirus: Aichivirus A (AiV-A) includes human AiV-1, canine KoV-1, and murine KoV-1. Aichivirus B (AiV-B) includes bovine KoV-1 and sheep KoV-1. Aichivirus C (AiV-C) includes porcine KoV-1 (PKoV/AiV-C).1 Cleaning and Disinfection • AiV-1 is readily inactivated at 56ºC after 20 minutes. • There is no published information about the susceptibility of PKoV/AiV-C to disinfectants. Kobuviruses (KoVs) are potentially susceptible to disinfection with acids like acetic acid, aldehydes like glutaraldehyde, alkalis like sodium hydroxide, and oxidizing agents like Virkon- S®11. Epidemiology • Kobuviruses infect many different species. The AiV-C cluster contains swine viruses exclusively. • PKoV/AiV-C has been isolated from swine herds in China, Thailand, Japan, South Korea, Italy, Hungary, Czech Republic, the United States, the Netherlands, Kenya, Uganda, and Brazil. • Prevalence in domestic pigs ranges from 13–99%. One study of pigs in the United States showed that 21.7% of healthy and 21.9% of diarrheic samples were PKoV/AiV-C-positive. Transmission • Transmission is thought to be fecal-oral. • Wild boars might be a source of infection for domestic swine. Infection in Swine/Pathogenesis • PKoV/AiV-C has been implicated as the cause of an outbreak of diarrhea, dehydration, and vomiting in Chinese piglets. -

Viral Diversity in Tree Species
Universidade de Brasília Instituto de Ciências Biológicas Departamento de Fitopatologia Programa de Pós-Graduação em Biologia Microbiana Doctoral Thesis Viral diversity in tree species FLÁVIA MILENE BARROS NERY Brasília - DF, 2020 FLÁVIA MILENE BARROS NERY Viral diversity in tree species Thesis presented to the University of Brasília as a partial requirement for obtaining the title of Doctor in Microbiology by the Post - Graduate Program in Microbiology. Advisor Dra. Rita de Cássia Pereira Carvalho Co-advisor Dr. Fernando Lucas Melo BRASÍLIA, DF - BRAZIL FICHA CATALOGRÁFICA NERY, F.M.B Viral diversity in tree species Flávia Milene Barros Nery Brasília, 2025 Pages number: 126 Doctoral Thesis - Programa de Pós-Graduação em Biologia Microbiana, Universidade de Brasília, DF. I - Virus, tree species, metagenomics, High-throughput sequencing II - Universidade de Brasília, PPBM/ IB III - Viral diversity in tree species A minha mãe Ruth Ao meu noivo Neil Dedico Agradecimentos A Deus, gratidão por tudo e por ter me dado uma família e amigos que me amam e me apoiam em todas as minhas escolhas. Minha mãe Ruth e meu noivo Neil por todo o apoio e cuidado durante os momentos mais difíceis que enfrentei durante minha jornada. Aos meus irmãos André, Diego e meu sobrinho Bruno Kawai, gratidão. Aos meus amigos de longa data Rafaelle, Evanessa, Chênia, Tati, Leo, Suzi, Camilets, Ricardito, Jorgito e Diego, saudade da nossa amizade e dos bons tempos. Amo vocês com todo o meu coração! Minha orientadora e grande amiga Profa Rita de Cássia Pereira Carvalho, a quem escolhi e fui escolhida para amar e fazer parte da família. -
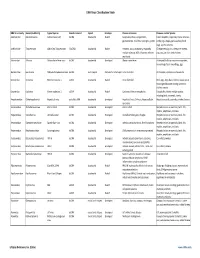
Virus Classification Tables V2.Vd.Xlsx
DNA Virus Classification Table DNA Virus Family Genera (Subfamily) Typical Species Genetic material Capsid Envelope Disease in Humans Diseases in other Species Adenoviridae Mastadenovirus Adenoviruses 1‐47 dsDNA Icosahedral Naked Respiratory illness; conjunctivitis, Canine hepatitis, respiratory illness in horses, gastroenteritis, tonsillitis, meningitis, cystitis cattle, pigs, sheep, goats, sea lions, birds dogs, squirrel enteritis Anelloviridae Torqueviruses Alpha‐Zeta Torqueviruses (‐)ssDNA Icosahedral Naked Hepatitis, lupus, pulmonary, myopathy, Chimpanzee, pig, cow, sheep, tree shrews, multiple sclerosis; 90% of humans infected pigs, cats, sea lions and chickens worldwide Asfarviridae Asfivirus African Swine fever virus dsDNA Icosahedral Enveloped African swine fever Arthropod (tick) transmission or ingestion; hemorrhagic fever in warthogs, pigs Baculoviridae Baculovirus Alpha‐Gamma Baculoviruses dsDNA Stick shaped Occluded or Enveloped none identified Arthropods, Lepidoptera, crustaceans Circoviridae Circovirus Porcine circovirus 1 ssDNA Icosahedral Naked none identified Birds, pigs, dogs; bats; rodents; causes post‐ weaning multisystem wasting syndrome, chicken anemia Circoviridae Cyclovirus Human cyclovirus 1 ssDNA Icosahedral Naked Cyclovirus Vietnam encephalitis Encephalitis; infects multiple species including birds, mammals, insects Hepadnaviridae Orthohepadnavirus Hepatitis B virus partially ssDNA Icosahedral Enveloped Hepatitis B virus; Cirrhosis, Hepatocellular Hepatitis in ducks, squirrels, primates, herons carcinoma Herpesviridae -

Viruses in Transplantation - Not Always Enemies
Viruses in transplantation - not always enemies Virome and transplantation ECCMID 2018 - Madrid Prof. Laurent Kaiser Head Division of Infectious Diseases Laboratory of Virology Geneva Center for Emerging Viral Diseases University Hospital of Geneva ESCMID eLibrary © by author Conflict of interest None ESCMID eLibrary © by author The human virome: definition? Repertoire of viruses found on the surface of/inside any body fluid/tissue • Eukaryotic DNA and RNA viruses • Prokaryotic DNA and RNA viruses (phages) 25 • The “main” viral community (up to 10 bacteriophages in humans) Haynes M. 2011, Metagenomic of the human body • Endogenous viral elements integrated into host chromosomes (8% of the human genome) • NGS is shaping the definition Rascovan N et al. Annu Rev Microbiol 2016;70:125-41 Popgeorgiev N et al. Intervirology 2013;56:395-412 Norman JM et al. Cell 2015;160:447-60 ESCMID eLibraryFoxman EF et al. Nat Rev Microbiol 2011;9:254-64 © by author Viruses routinely known to cause diseases (non exhaustive) Upper resp./oropharyngeal HSV 1 Influenza CNS Mumps virus Rhinovirus JC virus RSV Eye Herpes viruses Parainfluenza HSV Measles Coronavirus Adenovirus LCM virus Cytomegalovirus Flaviviruses Rabies HHV6 Poliovirus Heart Lower respiratory HTLV-1 Coxsackie B virus Rhinoviruses Parainfluenza virus HIV Coronaviruses Respiratory syncytial virus Parainfluenza virus Adenovirus Respiratory syncytial virus Coronaviruses Gastro-intestinal Influenza virus type A and B Human Bocavirus 1 Adenovirus Hepatitis virus type A, B, C, D, E Those that cause