And Type the TITLE of YOUR WORK in All Caps
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Summary of Offerings in the PBS Bulb Exchange, Dec 2012- Nov 2019
Summary of offerings in the PBS Bulb Exchange, Dec 2012- Nov 2019 3841 Number of items in BX 301 thru BX 463 1815 Number of unique text strings used as taxa 990 Taxa offered as bulbs 1056 Taxa offered as seeds 308 Number of genera This does not include the SXs. Top 20 Most Oft Listed: BULBS Times listed SEEDS Times listed Oxalis obtusa 53 Zephyranthes primulina 20 Oxalis flava 36 Rhodophiala bifida 14 Oxalis hirta 25 Habranthus tubispathus 13 Oxalis bowiei 22 Moraea villosa 13 Ferraria crispa 20 Veltheimia bracteata 13 Oxalis sp. 20 Clivia miniata 12 Oxalis purpurea 18 Zephyranthes drummondii 12 Lachenalia mutabilis 17 Zephyranthes reginae 11 Moraea sp. 17 Amaryllis belladonna 10 Amaryllis belladonna 14 Calochortus venustus 10 Oxalis luteola 14 Zephyranthes fosteri 10 Albuca sp. 13 Calochortus luteus 9 Moraea villosa 13 Crinum bulbispermum 9 Oxalis caprina 13 Habranthus robustus 9 Oxalis imbricata 12 Haemanthus albiflos 9 Oxalis namaquana 12 Nerine bowdenii 9 Oxalis engleriana 11 Cyclamen graecum 8 Oxalis melanosticta 'Ken Aslet'11 Fritillaria affinis 8 Moraea ciliata 10 Habranthus brachyandrus 8 Oxalis commutata 10 Zephyranthes 'Pink Beauty' 8 Summary of offerings in the PBS Bulb Exchange, Dec 2012- Nov 2019 Most taxa specify to species level. 34 taxa were listed as Genus sp. for bulbs 23 taxa were listed as Genus sp. for seeds 141 taxa were listed with quoted 'Variety' Top 20 Most often listed Genera BULBS SEEDS Genus N items BXs Genus N items BXs Oxalis 450 64 Zephyranthes 202 35 Lachenalia 125 47 Calochortus 94 15 Moraea 99 31 Moraea -

Approved Plant List 10/04/12
FLORIDA The best time to plant a tree is 20 years ago, the second best time to plant a tree is today. City of Sunrise Approved Plant List 10/04/12 Appendix A 10/4/12 APPROVED PLANT LIST FOR SINGLE FAMILY HOMES SG xx Slow Growing “xx” = minimum height in Small Mature tree height of less than 20 feet at time of planting feet OH Trees adjacent to overhead power lines Medium Mature tree height of between 21 – 40 feet U Trees within Utility Easements Large Mature tree height greater than 41 N Not acceptable for use as a replacement feet * Native Florida Species Varies Mature tree height depends on variety Mature size information based on Betrock’s Florida Landscape Plants Published 2001 GROUP “A” TREES Common Name Botanical Name Uses Mature Tree Size Avocado Persea Americana L Bahama Strongbark Bourreria orata * U, SG 6 S Bald Cypress Taxodium distichum * L Black Olive Shady Bucida buceras ‘Shady Lady’ L Lady Black Olive Bucida buceras L Brazil Beautyleaf Calophyllum brasiliense L Blolly Guapira discolor* M Bridalveil Tree Caesalpinia granadillo M Bulnesia Bulnesia arboria M Cinnecord Acacia choriophylla * U, SG 6 S Group ‘A’ Plant List for Single Family Homes Common Name Botanical Name Uses Mature Tree Size Citrus: Lemon, Citrus spp. OH S (except orange, Lime ect. Grapefruit) Citrus: Grapefruit Citrus paradisi M Trees Copperpod Peltophorum pterocarpum L Fiddlewood Citharexylum fruticosum * U, SG 8 S Floss Silk Tree Chorisia speciosa L Golden – Shower Cassia fistula L Green Buttonwood Conocarpus erectus * L Gumbo Limbo Bursera simaruba * L -
Additional Information
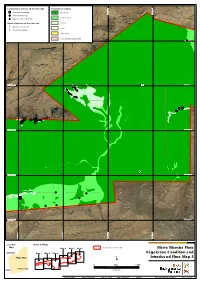
Current Survey Introduced Flora Records Vegetation Condition *Acetosa vesicaria Excellent 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 mE 535,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 536,000 mE 536,000 537,000 mE 537,000 534,000 mE 534,000 mE 535,000 534,000 mE 534,000 -

DNA Barcoding Identification of Endangered Dipcadi Saxorum Blatt
Human Journals Research Article June 2019 Vol.:15, Issue:3 © All rights are reserved by Purohit Nikisha et al. DNA Barcoding Identification of Endangered Dipcadi saxorum Blatt. Species of Gujarat Keywords: DNA Barcoding, rbcL, endangered plants, identification, Phylogenetic tree, conservation ABSTRACT Purohit Nikisha*1, Solanki Hiteshkumar A2. DNA barcoding is an appropriate molecular method which uses a short sequence as a barcoding region precise for identified species. It has the capability to fast the discovery of new [1]. Research Scholar, Department of Botany, Gujarat species. In this study, the potential of DNA barcoding to University, Ahmedabad-380009. approve the identities of endangered plant species in Dediyapada, Gujarat was assessed using DNA barcode rbcL. [2]. Professor, Department of Botany, Gujarat rbcL marker was successful in amplifying target regions for University, Ahmedabad-380009. Dipcadi saxorum Blatt. species. RbcL primer resulted in cleanest reads. Correct identification of any plant is a complete requirement. DNA barcoding is a reliable tool in methodically Submission: 26 May 2019 identifying unknown endangered plants. The current study Accepted: 31 May 2019 explains how DNA barcode analysis of the plant Dipcadi Published: 30 June 2019 saxorum Blatt. helps in the correct identification based on nucleotide diversity of short DNA segments. DNA from the leaf of the plant were extracted. The chloroplast gene rbcL were amplified by PCR and sequenced. The sequence was subjected to a BLAST analysis to compare it with that of other species and a phylogenetic tree was constructed. The results established that the plant belonged to the family Asparagaceae. Overall, the www.ijppr.humanjournals.com endangered species were precisely identified to the species level. -

UNIVERSITY of CALIFORNIA Santa Barbara Ancient Plant Use and the Importance of Geophytes Among the Island Chumash of Santa Cruz
UNIVERSITY OF CALIFORNIA Santa Barbara Ancient Plant Use and the Importance of Geophytes among the Island Chumash of Santa Cruz Island, California A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Anthropology by Kristina Marie Gill Committee in charge: Professor Michael A. Glassow, Chair Professor Michael A. Jochim Professor Amber M. VanDerwarker Professor Lynn H. Gamble September 2015 The dissertation of Kristina Marie Gill is approved. __________________________________________ Michael A. Jochim __________________________________________ Amber M. VanDerwarker __________________________________________ Lynn H. Gamble __________________________________________ Michael A. Glassow, Committee Chair July 2015 Ancient Plant Use and the Importance of Geophytes among the Island Chumash of Santa Cruz Island, California Copyright © 2015 By Kristina Marie Gill iii DEDICATION This dissertation is dedicated to my Family, Mike Glassow, and the Chumash People. iv ACKNOWLEDGEMENTS I am indebted to many people who have provided guidance, encouragement, and support in my career as an archaeologist, and especially through my undergraduate and graduate studies. For those of whom I am unable to personally thank here, know that I deeply appreciate your support. First and foremost, I want to thank my chair Michael Glassow for his patience, enthusiasm, and encouragement during all aspects of this daunting project. I am also truly grateful to have had the opportunity to know, learn from, and work with my other committee members, Mike Jochim, Amber VanDerwarker, and Lynn Gamble. I cherish my various field experiences with them all on the Channel Islands and especially in southern Germany with Mike Jochim, whose worldly perspective I value deeply. I also thank Terry Jones, who provided me many undergraduate opportunities in California archaeology and encouraged me to attend a field school on San Clemente Island with Mark Raab and Andy Yatsko, an experience that left me captivated with the islands and their history. -

Outline of Angiosperm Phylogeny
Outline of angiosperm phylogeny: orders, families, and representative genera with emphasis on Oregon native plants Priscilla Spears December 2013 The following listing gives an introduction to the phylogenetic classification of the flowering plants that has emerged in recent decades, and which is based on nucleic acid sequences as well as morphological and developmental data. This listing emphasizes temperate families of the Northern Hemisphere and is meant as an overview with examples of Oregon native plants. It includes many exotic genera that are grown in Oregon as ornamentals plus other plants of interest worldwide. The genera that are Oregon natives are printed in a blue font. Genera that are exotics are shown in black, however genera in blue may also contain non-native species. Names separated by a slash are alternatives or else the nomenclature is in flux. When several genera have the same common name, the names are separated by commas. The order of the family names is from the linear listing of families in the APG III report. For further information, see the references on the last page. Basal Angiosperms (ANITA grade) Amborellales Amborellaceae, sole family, the earliest branch of flowering plants, a shrub native to New Caledonia – Amborella Nymphaeales Hydatellaceae – aquatics from Australasia, previously classified as a grass Cabombaceae (water shield – Brasenia, fanwort – Cabomba) Nymphaeaceae (water lilies – Nymphaea; pond lilies – Nuphar) Austrobaileyales Schisandraceae (wild sarsaparilla, star vine – Schisandra; Japanese -

Wiregrass (Aristida Stricta)
Wiregrass (Aristida stricta) For definitions of botanical terms, visit en.wikipedia.org/wiki/Glossary_of_botanical_terms. Wiregrass is a perennial bunchgrass found in scrub, pinelands and coastal uplands throughout much of Florida. It is the dominant groundcover species in longleaf pine savannas and is a primary food source for gopher tortoises. Birds and small wildlife eat the seeds. Historically, cattle grazed on Wiregrass’s tender new growth. Wiregrass flowers are tiny and brown. They are born on spikelike terminal panicles Flower stalks are elongated and extend above the leaves. Leaf blades are long, thin and rolled inward, giving them a wiry appearance (hence the common name). They are erect and green when young Photo courtesy of Alan Cressler, Lady Bird Johnson Wildflower Center and begin to arch and turn brown as they age. Its fruits are small, yellowish caryopses. Seeds may be dispersed by wind, gravity or on the fur of passing animals. The genus name Aristida is from the Latin arista, meaning “awn” and referring to the three awns or bristle-like structures that extend from the florets. (An alternative common name is Pineland threeawn.) The species epithet, stricta, is from the Latin strictus, meaning straight or erect. Family: Poaceae (Grass family) Native range: Nearly throughout To see where natural populations of Wiregrass have been vouchered, visit www.florida.plantatlas.usf.edu. Hardiness: Zones 8A–10B Lifespan: Perennial Soil: Moist to dry, well-drained sandy soils Exposure: Full sun to partial shade Growth habit: 1–3’+ tall and equally wide Propagation: Division, seed Garden tips: Wiregrass is fast-growing and tolerant of drought conditions and low-nutrient soils. -

Flavonoid, Phenylethanoid and Iridoid Glycosides from Globularia Aphyllanthes
Flavonoid, Phenylethanoid and Iridoid Glycosides from Globularia aphyllanthes Hasan Kırmızıbekmeza, Carla Bassarellob, Sonia Piacenteb, Galip Akaydınc, and Ihsan˙ C¸ alıs¸d,e a Yeditepe University, Faculty of Pharmacy, Department of Pharmacognosy, TR-34755, Kayıs¸da˘gı, Istanbul,˙ Turkey b Salerno University, Department of Pharmaceutical Sciences, Via Ponte Don Melillo, 84084 Fisciano, Salerno, Italy c Hacettepe University, Department of Biology Education, TR-06800, Beytepe, Ankara, Turkey d Hacettepe University, Faculty of Pharmacy, Department of Pharmacognosy, TR-06100, Ankara, Turkey e Present address: Near East University, Faculty of Pharmacy, Department of Pharmacognosy and Pharmaceutical Botany, Nicosia, Turkish Republic of Northern Cyprus Reprint requests to Dr. Hasan Kırmızıbekmez. Fax: +90 216 578 00 68. E-mail: [email protected] Z. Naturforsch. 2009, 64b, 252 – 256; received September 4, 2008 A new flavone glycoside, 6-hydroxyluteolin 7-O-[6 -benzoyl-β-D-glucopyranosyl-(1 → 2)]-β- D-glucopyranoside (aphyllanthoside, 1) was isolated from the MeOH extract of the aerial parts of Globularia aphyllanthes. Besides this new compound, two flavonoid glycosides (6-hydroxyluteolin 7-O-[6 -(E)-caffeoyl-β-D-glucopyranosyl-(1 → 2)]-β-D-glucopyranoside and isoquercitrin), three phenylethanoid glycosides (verbascoside, rossicaside A, and trichosanthoside A), and 11 iridoid gly- cosides (aucubin, catalpol, 10-O-benzoylcatalpol, globularin, asperuloside, besperuloside, asperulo- sidic acid, daphylloside, scandoside, alpinoside and baldaccioside) were also obtained and character- ized. Identification of the isolated compounds was carried out by spectroscopic analysis including 1D and 2D NMR experiments as well as HRMS. Key words: Globularia aphyllanthes, Plantaginaceae, Flavonoids, Aphyllanthoside, Phenylethanoid and Iridoid Glycosides Introduction Experimental Section Globularia aphyllanthes Crantz (Plantaginaceae) is General experimental procedures a perennial plant, distributed from northwest Turkey to central and southern Europe [1]. -

Phd Federica Gilardelli A5
Vegetation dynamics and restoration trials in limestone quarries: the Botticino case study (Brescia, Italy) Federica Gilardelli UNIVERSITÀ DEGLI STUDI DI MILANO – BICOCCA Facoltà di Scienze Matematiche, Fisiche e Naturali Vegetation dynamics and restoration trials in limestone quarries: the Botticino case study (Brescia, Italy) Federica Gilardelli PhD thesis in Environmental Science XXV cycle Tutor: Cotutors: Prof. Sandra Citterio Prof. Sergio Sgorbati Dr. Rodolfo Gentili Dr. Stefano Armiraglio Collaborations: Dr. Ing. Sergio Savoldi Dr. Pierangelo Barossi February 2013 To all the quarrymen and their families. A tutti i cavatori e le loro famiglie. Background. All over the world, the naturalistic restoration of abandoned quarry areas represents a real challenge because of the very adverse initial site conditions for plant species colonization. In order to identify the best restoration practices, the present thesis considered, as a case study, the “Botticino extractive basin” (Lombardy, Italy), that is today the second greatest Italian extractive basin and it is famous worldwide for the limestone extraction. In particular, the thesis proposes a multidisciplinary approach based on the study of the local vegetation dynamics, laboratory tests, plant selection for restoration and field experiments to test different restoration techniques. Methods. Spontaneous vegetation dynamics over the whole extractive basin was studied by an ecological approach through 108 plots, that were carried out on surfaces whose “disused time” from quarry abandonment was known; data were analysed by cluster analysis and Canonical Correspondence Analysis (CCA) and compared to the available data on grassland and woodlands related to the study area. We identified successional phases according to the trend of the most common species whose cover significantly increases or decreases with time. -

Complete Chloroplast Genomes Shed Light on Phylogenetic
www.nature.com/scientificreports OPEN Complete chloroplast genomes shed light on phylogenetic relationships, divergence time, and biogeography of Allioideae (Amaryllidaceae) Ju Namgung1,4, Hoang Dang Khoa Do1,2,4, Changkyun Kim1, Hyeok Jae Choi3 & Joo‑Hwan Kim1* Allioideae includes economically important bulb crops such as garlic, onion, leeks, and some ornamental plants in Amaryllidaceae. Here, we reported the complete chloroplast genome (cpDNA) sequences of 17 species of Allioideae, fve of Amaryllidoideae, and one of Agapanthoideae. These cpDNA sequences represent 80 protein‑coding, 30 tRNA, and four rRNA genes, and range from 151,808 to 159,998 bp in length. Loss and pseudogenization of multiple genes (i.e., rps2, infA, and rpl22) appear to have occurred multiple times during the evolution of Alloideae. Additionally, eight mutation hotspots, including rps15-ycf1, rps16-trnQ-UUG, petG-trnW-CCA , psbA upstream, rpl32- trnL-UAG , ycf1, rpl22, matK, and ndhF, were identifed in the studied Allium species. Additionally, we present the frst phylogenomic analysis among the four tribes of Allioideae based on 74 cpDNA coding regions of 21 species of Allioideae, fve species of Amaryllidoideae, one species of Agapanthoideae, and fve species representing selected members of Asparagales. Our molecular phylogenomic results strongly support the monophyly of Allioideae, which is sister to Amaryllioideae. Within Allioideae, Tulbaghieae was sister to Gilliesieae‑Leucocoryneae whereas Allieae was sister to the clade of Tulbaghieae‑ Gilliesieae‑Leucocoryneae. Molecular dating analyses revealed the crown age of Allioideae in the Eocene (40.1 mya) followed by diferentiation of Allieae in the early Miocene (21.3 mya). The split of Gilliesieae from Leucocoryneae was estimated at 16.5 mya. -

Dyuhei Sato Division of Genetics, Bot. Inst. Faculty of Science, Tokyo
ANALYSIS OF THE KARYOTYPES IN YUCCA, A GA VE AND THE RELATED GENERA WITH SPECIAL REFERENCE TO THE PHYLOGENETIC SIGNIFICANCEI~ Dyuhei SATo Divisionof Genetics, Bot. Inst. Faculty of Science, Tokyo Imperial University McKelvey and Sax (2933) have called attention to the existence of taxonomic and cytological similarities of the genera Yucca, Hesperoyucca, Gleistvucca,Hesperoaloe and Samuela of the Liliaceae with the genera Agave and Fourcroya which belong to a related family, Amaryllidaceae. Wh.itaker (1934) also has reported that Polianhes and Fourcroya have exactly the same chromosome constitution as the Yucca-Abave karyotype (5 long and 25 short chromosomes) (Figs. 1, 2). These observations when considered in respect to taxonomic resemblances, seem to indicate that the genera mentioned above are more closely related than it is shown by their classifica- tion into distinct families. Whitaker also has remarked that Dasylirion (2n=38) and ATolina(2n=36) in Yucceae and Doryanthes (2n=36) in Agavoideae are of different karyotypes from the Yucca-Agave type. In the present work an analysis of the karyotypes in Liliaceous plants has been attempted and several karyotypes have been found in Scilloideae. Eucornis and Carassia have been selected with the purpose of discovering a possible connecting link between these genera and the Yucca-Agave group. In the present paper an analysis of the karyotypes of the following species is given. LILIACEAE Scilloideae 211 Fig. Euconis undulata 60=8L+8M+44S (4b)2) 3 Euconsispallidi ora 60=8L+8M+44S (4b) 4 Eucomispunctata 60=8L±8M+44S (4b) 5 Camassiaescrema 30=6L+24S (2b) 6 Yucceae Yuccafilamentosa 30 60=1OL+50S (2b) 1, 7 Yuccarecurvifolia 30 60=1OL+50S (2b) 2, 8 Yuccaaloifolia 60=1OL+50S (2b) 9 „ var. -

Evolutionary History of Floral Key Innovations in Angiosperms Elisabeth Reyes
Evolutionary history of floral key innovations in angiosperms Elisabeth Reyes To cite this version: Elisabeth Reyes. Evolutionary history of floral key innovations in angiosperms. Botanics. Université Paris Saclay (COmUE), 2016. English. NNT : 2016SACLS489. tel-01443353 HAL Id: tel-01443353 https://tel.archives-ouvertes.fr/tel-01443353 Submitted on 23 Jan 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. NNT : 2016SACLS489 THESE DE DOCTORAT DE L’UNIVERSITE PARIS-SACLAY, préparée à l’Université Paris-Sud ÉCOLE DOCTORALE N° 567 Sciences du Végétal : du Gène à l’Ecosystème Spécialité de Doctorat : Biologie Par Mme Elisabeth Reyes Evolutionary history of floral key innovations in angiosperms Thèse présentée et soutenue à Orsay, le 13 décembre 2016 : Composition du Jury : M. Ronse de Craene, Louis Directeur de recherche aux Jardins Rapporteur Botaniques Royaux d’Édimbourg M. Forest, Félix Directeur de recherche aux Jardins Rapporteur Botaniques Royaux de Kew Mme. Damerval, Catherine Directrice de recherche au Moulon Président du jury M. Lowry, Porter Curateur en chef aux Jardins Examinateur Botaniques du Missouri M. Haevermans, Thomas Maître de conférences au MNHN Examinateur Mme. Nadot, Sophie Professeur à l’Université Paris-Sud Directeur de thèse M.