Draft Genome Sequence of Bacillus Azotoformans MEV2011, a (Co
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Heterotrophic Denitrification by Gram-Positive Bacteria: Bacillus Cereus and Bacillus Tequilensis
International Journal of Scientific and Research Publications, Volume 4, Issue 4, April 2014 1 ISSN 2250-3153 Heterotrophic denitrification by Gram-positive bacteria: Bacillus cereus and Bacillus tequilensis Moukhlissi Saïd*, Aboussabiq Fatima Ezzahra*, Amine Jamal*, Rihani Mohammed* and Assobhei Omar* * Laboratory of Marine Biotechnology and Environment, Faculty of Science of El Jadida, P.O. Box 20, El Jadida 24000, Morocco. Abstract- Two bacteria were isolated from anoxic denitrifying notoriously overlooked in community analysis of denitrifiers in reactor for treatment of domestic wastewater. The analysis of the the environment because they are not targeted by the available 16S rDNA gene sequences showed that the isolated strains were PCR primers designed for denitrification genes (Throbäck et al., affiliated with Bacillus cereus and Bacillus tequilensis. 2004). Verbaendert et al. (2011) have studied the denitrification Denitrification was compared between Bacillus cereus and of a large collection of Bacillus strains and suggested that Bacillus tequilensis in this study. Two bacilli were able to denitrification occurred in nearly half of the analysed strains. denitrify and Bacillus cereus was more efficient than Bacillus More recently, a variety of bacilli were tested for gas production tequilensis. Bacillus cereus reduced 80% of high amount of under denitrifying conditions and found to be complete nitrate; however, Bacillus tequilensis could reduce 37.4% of denitrifiers (Jones et al., 2011). Genome sequencing has revealed nitrate. These heterotrophic bacteria are able to eliminate organic the potential for partial denitrification in some Bacillus species. matter with the same trend reducing 74.5% for Bacillus For example, qNor is present in Bacillus tusciae strain DSM2912 tequilensis and 70.2% for Bacillus cereus. -

Identification and Characterization of Bacillus Cereus
Journal of Insect Science RESEARCH Identification and Characterization of Bacillus cereus SW7-1 in Bombyx mori (Lepidoptera: Bombycidae) Guan-Nan Li,1,2 Xue-Juan Xia,3 Huan-Huan Zhao,1,2 Parfait Sendegeya,1,2 and Yong Zhu1,2,4 1College of Biotechnology, Southwest University, Chongqing 400716, China 2State Key Laboratory of Silkworm Genome Biology, Chongqing 400716, China 3College of Food Science, Southwest University, Chongqing 400716, China 4Corresponding author, e-mail: [email protected] Subject Editor: Seth Barribeau J. Insect Sci. (2015) 15(1): 136; DOI: 10.1093/jisesa/iev121 ABSTRACT. The bacterial diseases of silkworms cause significant reductions in sericulture and result in huge economic loss. This study aimed to identify and characterize a pathogen from diseased silkworm. SW7-1, a pathogenic bacterial strain, was isolated from the dis- eased silkworm. The strain was identified on the basis of its bacteriological properties and 16S rRNA gene sequence. The colony was round, slightly convex, opaque, dry, and milky on a nutrient agar medium, the colony also exhibited jagged edges. SW7-1 was Gram- positive, without parasporal crystal, and 0.8–1.2 by 2.6–3.4 mm in length, resembling long rods with rounded ends. The strain was posi- tive to most of the physiological biochemical tests used in this study. The strain could utilize glucose, sucrose, and maltose. The results of its 16S rRNA gene sequence analysis revealed that SW7-1 shared the highest sequence identity (>99%) with Bacillus cereus strain 14. The bacterial strain was highly susceptible to gentamycin, streptomycin, erythromycin, norfloxacin, and ofloxacin and moderately susceptible to tetracycline and rifampicin. -
![Downloaded from the Microbial Genome (Complete and Non-Classical Protein Secretion [73]](https://docslib.b-cdn.net/cover/5140/downloaded-from-the-microbial-genome-complete-and-non-classical-protein-secretion-73-3625140.webp)
Downloaded from the Microbial Genome (Complete and Non-Classical Protein Secretion [73]
Helen et al. BMC Genomics (2016) 17:155 DOI 10.1186/s12864-016-2465-0 RESEARCH ARTICLE Open Access Highly diverse nirK genes comprise two major clades that harbour ammonium- producing denitrifiers Decleyre Helen, Heylen Kim*, Bjorn Tytgat and Willems Anne* Abstract Background: Copper dependent nitrite reductase, NirK, catalyses the key step in denitrification, i.e. nitrite reduction to nitric oxide. Distinct structural NirK classes and phylogenetic clades of NirK-type denitrifiers have previously been observed based on a limited set of NirK sequences, however, their environmental distribution or ecological strategies are currently unknown. In addition, environmental nirK-type denitrifiers are currently underestimated in PCR-dependent surveys due to primer coverage limitations that can be attributed to their broad taxonomic diversity and enormous nirK sequence divergence. Therefore, we revisited reported analyses on partial NirK sequences using a taxonomically diverse, full-length NirK sequence dataset. Results: Division of NirK sequences into two phylogenetically distinct clades was confirmed, with Clade I mainly comprising Alphaproteobacteria (plus some Gamma- and Betaproteobacteria) and Clade II harbouring more diverse taxonomic groups like Archaea, Bacteroidetes, Chloroflexi, Gemmatimonadetes, Nitrospirae, Firmicutes, Actinobacteria, Planctomycetes and Proteobacteria (mainly Beta and Gamma). Failure of currently available primer sets to target diverse NirK-type denitrifiers in environmental surveys could be attributed to mismatches over the whole length of the primer binding regions including the 3′ site, with Clade II sequences containing higher sequence divergence than Clade I sequences. Simultaneous presence of both the denitrification and DNRA pathway could be observed in 67 % of all NirK-type denitrifiers. Conclusion: The previously reported division of NirK into two distinct phylogenetic clades was confirmed using a taxonomically diverse set of full-length NirK sequences. -
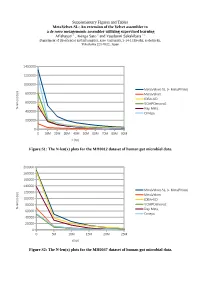
Supplementary Figures and Tables Metavelvet-SL
Supplementary Figures and Tables MetaVelvet-SL: An extension of the Velvet assembler to a de novo metagenomic assembler utilizing supervised learning Afiahayati 1 , Kengo Sato 1 and Yasubumi Sakakibara 1∗ Department of Biosciences and Informatics, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan 1400000 1200000 1000000 MetaVelvet-SL (+ MetaPhlAn) ) p 800000 b MetaVelvet ( ) x ( IDBA-UD n 600000 e SOAPDenovo2 l - N Ray Meta 400000 Omega 200000 0 0 10M 20M 30M 40M 50M 60M 70M 80M 90M x (bp) Figure S1: The N-len(x) plots for the MH0012 dataset of human gut microbial data. 200000 180000 160000 140000 MetaVelvet-SL (+ MetaPhlAn) ) 120000 p b MetaVelvet ( ) 100000 x IDBA-UD ( n e l 80000 SOAPDenovo2 - N Ray Meta 60000 Omega 40000 20000 0 0 5M 10M 15M 20M 25M x(bp) Figure S2: The N-len(x) plots for the MH0047 dataset of human gut microbial data. 600000 500000 400000 MetaVelvet-SL (+ MetaPhlAn) ) p b MetaVelvet ( ) 300000 x IDBA-UD ( n e l SOAPDenovo2 - N 200000 Ray Meta Omega 100000 0 0 10M 20M 30M 40M 50M 60M 70M 80M 90M x (bp) Figure S3: The N-len(x) plots for the SRS017227 dataset of human gut microbial data. 800000 700000 600000 500000 MetaVelvet-SL (+ MetaPhlAn) ) p b MetaVelvet ( ) 400000 x IDBA-UD ( n e l SOAPDenovo2 - 300000 N Ray Meta 200000 Omega 100000 0 0 5M 10M 15M 20M 25M 30M 35M 40M 45M x (bp) Figure S4: The N-len(x) plots for the SRS018661 dataset of human gut microbial data. Formula to identify unique nodes in Velvet (Zerbino and Birney, 2008) ̄x2 ρ2− log2 2 F ( ̄x ,n,ρ )= +n 2 2 where ̄x = the coverage of node ρ = expected coverage of subgraph n = the length of node A node is “unique”, if its F > 5 . -

Paenibacillus and Emended Description of the Genus Paenibacillus OSAMU SHIDA,H HIROAKI TAKAGI,L KIYOSHI KADOWAKI,L LAWRE CE K
7729 I 'TERNATIONAL JOURNAL OF SYSTEMATIC BACTERIOLOGY, Apr. 1997, p. 289-298 Vol. 47, 0.2 0020-7713/97/$04.00 0 Copyright © 1997, International Union of Microbiological Societies Transfer of Bacillus alginolyticus, Bacillus chondroitinus, Bacillus curdlanolyticus, Bacillus glucanolyticus, Bacillus kobensis, and Bacillus thiaminolyticus to the Genus Paenibacillus and Emended Description of the Genus Paenibacillus OSAMU SHIDA,h HIROAKI TAKAGI,l KIYOSHI KADOWAKI,l LAWRE CE K. NAKAMURA,2 3 AD KAZUO KOMAGATA Research Laboratory, Higeta Shoyu Co., Ltd., Choshi, Chiba 288,1 and Department ofAgricultural Chemistry, Faculty ofAgriculture, Tokyo University ofAgriculture, Setagaya-ku, Tokyo 156,3 Japan, and Microbial Properties Research, National Center for Agricultural Utilization Research, Us. Department ofAgriculture, Peoria, Illinois 616042 We determined the taxonomic status of six Bacillus species (Bacillus alginolyticus, Bacillus chondroitinus, Bacillus curdlanolyticus, Bacillus glucanolyticus, Bacillus kobensis, and Bacillus thiaminolyticus) by using the results of 168 rR A gene sequence and cellular fatty acid composition analyses. Phylogenetic analysis clus tered these species closely with the Paenibacillus species. Like the Paenibacillus species, the six Bacillus species contained anteiso-C 1s:o fatty acid as a major cellular fatty acid. The use of a specific PCR primer designed for differentiating the genus Paenibacillus from other members of the Bacillaceae showed that the six Bacillus species had the same amplified 168 rRNA gene fragment as members of the genus Paenibacillus. Based on these observations and other taxonomic characteristics, the six Bacillus species were transferred to the genus Paenibacillus. In addition, we propose emendation of the genus Paenibacillus. Rod-shaped, aerobic, endospore-forming bacteria have gen among the Bacillaceae, we determined the sequences of their erally been assigned to the genus Bacillus, a systematically 16S rR A genes and compared these sequences with homol diverse taxon (5). -

Bacillus Sp. Strain ZYK, a Selenite and Nitrate Reducer from Paddy Soil
Standards in Genomic Sciences (2014) 9:646-654 DOI:10.4056/sig s.3817480 Genome sequence of the anaerobic bacterium Bacillus sp. strain ZYK, a selenite and nitrate reducer from paddy soil PengBao1, Jian-QiangSu 2, Zheng -YiHu 3, Max M.Häggblom4, Yong -GuanZhu1,2* 1 State Key Lab of Urban and Regional Ecology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing, P. R. China 2 Key Lab of Urban Environment and Health, Institute of Urban Environment, Chinese Academy of Sciences, Xiamen, P. R. China 3 College of Resources and Environment, University of Chinese Academy of Sciences, Beijing , China 4 Rutgers University, Department of Biochemistry and Microbiology, School of Environmental and Biological Sciences, New Brunswick, NJ, USA Correspondence: Yong -Guan Zhu ([email protected]) Keywords: anaerobic, spore-forming , Gram-positive, nitrate-reduction, selenite-reduction, ar- senic resistance, paddy soil, Bacillaceae Bacillus sp. strain ZYK, a member of the phylum Firmicutes, is of interest for its ability to reduce nitrate and selenite and for its resistance to arsenic under anaerobic conditions. Here we de- scribe some key features of this organism, together with the complete genome sequence and annotation. The 3,575,797 bp long chromosome with its 3,454 protein-coding and 70 RNA genes, and the information gained from its sequence will be relevant to the elucidation of microbially-mediated transformations of nitrogen, selenium and arsenic in paddy soil. Introduction Classification and features Bacillus sp. ZYK (=DSM 26460 =CGMCC 1.5179) Based on 16S rRNA gene phylogeny and genome was isolated from a paddy soil in Dehong, Yunnan, information, strain ZYK was a member of the genus China and is an anaerobic nitrate-reducing, Gram- Bacillus, most closely related to Bacillus positive bacterium [1]. -

Large Scale Function-Based Genome Prospecting for Industrial Traits Applied to 1,3-Propanediol Production
bioRxiv preprint doi: https://doi.org/10.1101/2021.08.25.457110; this version posted August 27, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Large scale function-based genome prospecting for industrial traits applied to 1,3-propanediol production. Jasper J. Koehorst ID 1*, , Nikolaos Strepis ID 1,2*, Sanne de Graaf1, Alfons J. M. Stams ID 2,3, Diana Z. Sousa ID 2, and Peter J. Schaap ID 1 1Laboratory of Systems and Synthetic Biology | Department of Agrotechnology and Food Sciences, Wageningen University and Research, Wageningen, the Netherlands 2Laboratory of Microbiology | Department of Agrotechnology and Food Sciences, Wageningen University and Research, Wageningen, the Netherlands 3Centre of Biological Engineering, University of Minho, 4710-057 Braga, Portugal Due to the success of next-generation sequencing, there has been normally used. However, over larger phylogenetic distances, a vast build-up of microbial genomes in the public reposito- strategies that use sequence-based clustering algorithms are ries. FAIR genome prospecting of this huge genomic potential hampered by accumulation of evolutionary signals, lateral for biotechnological benefiting, require new efficient and flexible gene transfer, gene fusion/fission events and domain expan- methods. In this study, Semantic Web technologies are applied sions (5). Furthermore, when applied at larger scales, such to develop a function-based genome mining approach that fol- strategies suffer from high computational time and mem- lows a knowledge and discovery in database (KDD) protocol. -

COLETÂNEA DE PROCEDIMENTOS TÉCNICOS E METODOLOGIAS EMPREGADAS PARA O ESTUDO DE Bacillus E GÊNEROS ESPORULADOS AERÓBIOS CORRELATOS
Instituto Oswaldo Cruz Laboratório de Fisiologia Bacteriana COLETÂNEA DE PROCEDIMENTOS TÉCNICOS E METODOLOGIAS EMPREGADAS PARA O ESTUDO DE Bacillus E GÊNEROS ESPORULADOS AERÓBIOS CORRELATOS AUTORES Leon Rabinovitch Edmar Justo de Oliveira 2015 Laboratório de Fisiologia Bacteriana COLETÂNEA DE PROCEDIMENTOS TÉCNICOS E METODOLOGIAS EMPREGADAS PARA O ESTUDO DE Bacillus E GÊNEROS ESPORULADOS AERÓBIOS CORRELATOS Laboratório de Fisiologia Bacteriana – LFB (Coleção de Culturas do Gênero Bacillus e Gêneros Correlatos – CCGB e Laboratório de Referência Nacional para Carbúnculo – LARENAC) Laboratório de Fisiologia Bacteriana - LFB Coleção de Culturas do Gênero Bacillus e Gêneros Correlatos - CCGB Laboratório de Referência Nacional para Carbúnculo - LARENAC Tels: 2562-1640/2562-1637/2562-1639 Pavilhão Rocha Lima, 3º andar, salas 300-308-310-312 Av. Brasil, 4365, Manguinhos Rio de Janeiro – RJ CEP: 21040-900 e-mail: [email protected] Dados Internacionais de Catalogação-na-Publicação (CIP) Col683 Coletânea de procedimentos técnicos e metodologias empregadas para o estudo de Bacillus e gêneros esporulados aeróbios correlatos / Autores: Leon Rabinovitch e Edmar Justo de Oliveira. – 1. Ed. – Rio de Janeiro : Montenegro Comunicação, 2015. 160p. ; 21x28cm. Inclui bibliografia. ISBN 978-85-67506-04-3 (broch.) 1. Bacteriologia. I. Rabinovitch, Leon. II. Oliveira, Edmar Justo. CDD 610.8 Laboratório de Fisiologia Bacteriana COLETÂNEA DE PROCEDIMENTOS TÉCNICOS E METODOLOGIAS EMPREGADAS PARA O ESTUDO DE Bacillus E GÊNEROS ESPORULADOS AERÓBIOS CORRELATOS O Laboratório de Fisiologia Bacteriana, LFB, do Instituto Oswaldo Cruz, da Fundação Oswaldo Cruz é integrado por seto- res de pesquisa, ensino e desenvolvimento tecnológico, tendo na sua estrutura o Laboratório de Referência Nacional para Carbúnculo, LARENAC e Coleção de Culturas do Gênero Bacillus e Gêneros Correlatos, CCGB, filiada à Federação Mundial de Coleções de Culturas, WFCC. -

Proposal for Bacillus Smithii Sp. Nov
INTERNATIONAL JOURNAL OF SYSTEMATIC BACTERIOLOGY, Jan. 1988, p. 63-73 Vol. 38, No.1 0020-7713/88/010063-11$02.00/0 Taxonomic Study of Bacillus coagulans Hammer 1915 with a Proposal for Bacillus smithii sp. nov. LAWRENCE K. NAKAMURA,l* INGE BLUMENSTOCK,2 AND DIETER CLAUS2 Northern Regional Research Center, Agricultural Research Service, U.S. Department ofAgriculture, Peoria, Illinois 61604,1 and Deutsche Sammlung von Mikroorganismen, 3400 Gottingen, Federal Republic ofGermany2 Guanine-plus-cytosine (G+C) content determinations, deoxyribonucleic acid (DNA) relatedness estimations, and phenotypic similarity analyses of 90 strains identified previously as Bacillus coagulans Hammer 1915 revealed that 52 (group 1) strains were Bacillus coagulans sensu strfcto; 30 of the remaining organisms segregated into two distinct DNA relatedness groups, one consisting of 26 (group 2) strains and the other of 4 (group 4) organisms. Five (group 3) strains were Bacillus licheniformis, and three (group 5) strains were B. stearothermophilus. Because group 2 was a major cluster of strains which did not correspond in their characteristics to any previously known group, efforts were made to determine its taxonomic position. Group 2, with G+C contents ranging from 37 to 40 mol%, was compared with other species generally characterized as having G+C contents ranging from 37 to 44 mol% or capable of growing at 50°C or both (namely, Bacillus alcalophilus, Bacillus azotoformans, Bacillus badius, Bacillusfirmus, Bacillus globiformis, Bacillus laterosporus, Bacillus macquariensis, Bacillus marinus, Bacillus megaterium, Bacillus pumilus, and Bacillus subtilis). Low DNA relatedness values and poor matching of phenotypic characteristics strongly indicated that group 2 organisms are strains of a new species, for which the name Bacillus smithii is proposed. -

Qualitative and Quantitative Assessment of the “Dangerous
Center for International and Security Studies at Maryland1 Qualitative and Quantitative Assessment of the “Dangerous Activities” Categories Jens H. Kuhn July 2005 CISSM School of Public Policy This paper was prepared as part of the Advanced Methods of Cooperative Security Program at the Center 4113 Van Munching Hall for International and Security Studies at Maryland, with generous support from the MacArthur Foundation University of Maryland and the Sloan Foundation. College Park, MD 20742 Tel: (301) 405-7601 [email protected] 2 QUALITATIVE AND QUANTITATIVE ASSESSMENT OF THE “DANGEROUS ACTIVITIES” CATEGORIES DEFINED BY THE CISSM CONTROLLING DANGEROUS PATHOGENS PROJECT WORKING PAPER (July 31, 2005) Jens H. Kuhn, MD, ScD (Med. Sci.), MS (Biochem.) Contact Address: New England Primate Research Center Department of Microbiology and Molecular Genetics Harvard Medical School 1 Pine Hill Drive Southborough, MA 01772-9102, USA Phone: (508) 786-3326 Fax: (508) 786-3317 Email: [email protected] 3 OBJECTIVE The Controlling Dangerous Pathogens Project of the Center for International Security Studies at Maryland (CISSM) outlines a prototype oversight system for ongoing microbiological research to control its possible misapplication. This so-called Biological Research Security System (BRSS) foresees the creation of regional, national, and international oversight bodies that review, approve, or reject those proposed microbiological research projects that would fit three BRSS-defined categories: Potentially Dangerous Activities (PDA), Moderately Dangerous Activities (MDA), and Extremely Dangerous Activities (EDA). It is the objective of this working paper to assess these categories qualitatively and quantitatively. To do so, published US research of the years 2000-present (early- to mid-2005) will be screened for science reports that would have fallen under the proposed oversight system had it existed already. -

Dissimilatory Nitrate Reduction in Bacillus Copyright ©2017, Yihua Sun ISBN-Number: 978-94-6197-486-0 All Rights Are Reserved
Dissimilatory nitrate reduction in Bacillus Yihua Sun Promotors Dr. Kim Heylen Prof. Dr. em. Paul De Vos Prof. Dr. Anne Willems Dissertation submitted in fulfilment of the requirements for the degree of Doctor (Ph.D.) of Science: Biotechnology (Ghent University) Yihua Sun – Dissimilatory nitrate reduction in Bacillus Copyright ©2017, Yihua Sun ISBN-number: 978-94-6197-486-0 All rights are reserved. No part of this thesis protected by this copyright notice may be reproduced or utilized in any form or by any means, electronic or mechanical, including photocopying, recording or by any information storage or retrieval system without written permission of the author and promotors. Printed by University Press | www.universitypress.be Ph.D. thesis, Faculty of Sciences, Ghent University, Ghent, Belgium. This Ph.D. work was financially supported by Chinese scholar council ( 201206330054) and BOF CSC co- funding from Ghent University (01SC2713) Publicly defended in Ghent, Belgium, January 20 th , 2017 Examination committee Prof. Dr. Savvas Savvides (Chairman) L-Probe: Laboratory for Protein Biochemistry and Biomolecular Engineering Faculty of Sciences, Ghent University, Belgium VIB Inflammation Research Center VIB, Ghent, Belgium Prof. Dr. Anne Willems (Promotor) LM-UGent: Laboratory of Microbiology Faculty of Sciences, Ghent University, Belgium Prof. Dr. em. Paul De Vos (Promotor) LM-UGent: Laboratory of Microbiology Faculty of Sciences, Ghent University, Belgium Dr. Kim Heylen (Promotor) LM-UGent: Laboratory of Microbiology Faculty of Sciences, Ghent University, Belgium Prof. Dr. Sofie Goormachtig (Secretary) PSB: Plant systems Biology Faculty of Sciences, VIB, Ghent University, Belgium Prof. Dr. ir. Nico Boon CMET: Center for Microbial Ecology and Technology Faculty of Bioscience Engineering, Ghent University, Belgium Prof. -

Nitrous Oxide Emission by the Non- Denitrifying, Nitrate Ammonifier Bacillus Licheniformis
Sun et al. BMC Genomics (2016) 17:68 DOI 10.1186/s12864-016-2382-2 RESEARCH ARTICLE Open Access Nitrous oxide emission by the non- denitrifying, nitrate ammonifier Bacillus licheniformis Yihua Sun1, Paul De Vos1,2 and Kim Heylen1* Abstract Background: Firmicutes have the capacity to remove excess nitrate from the environment via either denitrification, dissimilatory nitrate reduction to ammonium or both. The recent renewed interest in their nitrogen metabolism has revealed many interesting features, the most striking being their wide variety of dissimilatory nitrate reduction pathways. In the present study, nitrous oxide production from Bacillus licheniformis, a ubiquitous Gram-positive, spore-forming species with many industrial applications, is investigated. Results: B. licheniformis has long been considered a denitrifier but physiological experiments on three different strains demonstrated that nitrous oxide is not produced from nitrate in stoichiometric amounts, rather ammonium is the most important end-product, produced during fermentation. Significant strain dependency in end-product ratios, attributed to nitrite and ammonium, and medium dependency in nitrous oxide production were also observed. Genome analyses confirmed the lack of a nitrite reductase to nitric oxide, the key enzyme of denitrification. Based on the gene inventory and building on knowledge from other non-denitrifying nitrous oxide emitters, hypothetical pathways for nitrous oxide production, involving NarG, NirB, qNor and Hmp, are proposed. In addition, all publically available genomes of B. licheniformis demonstrated similar gene inventories, with specific duplications of the nar operon, narK and hmp genes as well as NarG phylogeny supporting the evolutionary separation of previously described distinct BALI1 and BALI2 lineages. Conclusions: Using physiological and genomic data we have demonstrated that the common soil bacterium B.