1 This Manuscript Is Submitted for Publication in Renewable and Sustainable Energy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genome-Resolved Meta-Analysis of the Microbiome in Oil Reservoirs Worldwide
microorganisms Article Genome-Resolved Meta-Analysis of the Microbiome in Oil Reservoirs Worldwide Kelly J. Hidalgo 1,2,* , Isabel N. Sierra-Garcia 3 , German Zafra 4 and Valéria M. de Oliveira 1 1 Microbial Resources Division, Research Center for Chemistry, Biology and Agriculture (CPQBA), University of Campinas–UNICAMP, Av. Alexandre Cazellato 999, 13148-218 Paulínia, Brazil; [email protected] 2 Graduate Program in Genetics and Molecular Biology, Institute of Biology, University of Campinas (UNICAMP), Rua Monteiro Lobato 255, Cidade Universitária, 13083-862 Campinas, Brazil 3 Biology Department & CESAM, University of Aveiro, Aveiro, Portugal, Campus de Santiago, Avenida João Jacinto de Magalhães, 3810-193 Aveiro, Portugal; [email protected] 4 Grupo de Investigación en Bioquímica y Microbiología (GIBIM), Escuela de Microbiología, Universidad Industrial de Santander, Cra 27 calle 9, 680002 Bucaramanga, Colombia; [email protected] * Correspondence: [email protected]; Tel.: +55-19981721510 Abstract: Microorganisms inhabiting subsurface petroleum reservoirs are key players in biochemical transformations. The interactions of microbial communities in these environments are highly complex and still poorly understood. This work aimed to assess publicly available metagenomes from oil reservoirs and implement a robust pipeline of genome-resolved metagenomics to decipher metabolic and taxonomic profiles of petroleum reservoirs worldwide. Analysis of 301.2 Gb of metagenomic information derived from heavily flooded petroleum reservoirs in China and Alaska to non-flooded petroleum reservoirs in Brazil enabled us to reconstruct 148 metagenome-assembled genomes (MAGs) of high and medium quality. At the phylum level, 74% of MAGs belonged to bacteria and 26% to archaea. The profiles of these MAGs were related to the physicochemical parameters and recovery management applied. -

Genotyping of Uncultured Archaea in a Polluted Site of Suez Gulf, Egypt, Based on 16S Rrna Gene Analyses
Egyptian Journal of Aquatic Research (2014) 40,27–33 National Institute of Oceanography and Fisheries Egyptian Journal of Aquatic Research http://ees.elsevier.com/ejar www.sciencedirect.com FULL LENGTH ARTICLE Genotyping of uncultured archaea in a polluted site of Suez Gulf, Egypt, based on 16S rRNA gene analyses Hosam Easa Elsaied * Aquagenome Resources and Biotechnology Research Group, National Institute of Oceanography, 101-El-Kasr El-Eini Street, Cairo, Egypt Received 3 February 2014; revised 11 March 2014; accepted 11 March 2014 Available online 18 April 2014 KEYWORDS Abstract Culture-independent 16S rRNA gene analysis approach was used to explore and evalu- Archaea; ate archaea in a polluted site, El-Zeitia, Suez Gulf, Egypt. Metagenomic DNA was extracted from a 16S rRNA gene diversity; sediment sample. Archaeal 16S rRNA gene was PCR amplified using universal archaeal primers, Sediment; followed by cloning and direct analyses by sequencing. Rarefaction analysis showed saturation, Suez Gulf recording 21 archaeal 16S rRNA gene phylotypes, which represented the total composition of archaea in the studied sample. Phylogenetic analysis showed that all recorded phylotypes belonged to two archaeal phyla. Sixteen phylotypes were located in the branch of methanogenic Eur- yarchaeota and more closely related to species of the genera Methanosaeta and Methanomassiliicoc- cus. Five phylotypes were affiliated to the new archaeal phylum Thaumarchaeota, which represented by species Candidatus nitrosopumilus. The recorded phylotypes had unique sequences, characteriz- ing them as new phylogenetic lineages. This work is the first investigation of uncultured archaea in the Suez Gulf, and implicated that the environmental characteristics shaped the diversity of archa- eal 16S rRNA genes in the studied sample. -

Archaeoglobus Profundus Type Strain (AV18T)
Standards in Genomic Sciences (2010) 2:327-346 DOI:10.4056/sigs.942153 Complete genome sequence of Archaeoglobus profundus type strain (AV18T) Mathias von Jan1, Alla Lapidus2, Tijana Glavina Del Rio2, Alex Copeland2, Hope Tice2, Jan-Fang Cheng2, Susan Lucas2, Feng Chen2, Matt Nolan2, Lynne Goodwin2,3, Cliff Han2,3, Sam Pitluck2, Konstantinos Liolios2, Natalia Ivanova2, Konstantinos Mavromatis2, Galina Ovchinnikova2, Olga Chertkov2, Amrita Pati2, Amy Chen4, Krishna Palaniappan4, Miriam Land2,5, Loren Hauser2,5, Yun-Juan Chang2,5, Cynthia D. Jeffries2,5, Elizabeth Saunders2, Thomas Brettin2,3, John C. Detter2,3, Patrick Chain2,4, Konrad Eichinger6, Harald Huber6, Ste- fan Spring1, Manfred Rohde7, Markus Göker1, Reinhard Wirth6, Tanja Woyke2, Jim Bristow2, Jonathan A. Eisen2,8, Victor Markowitz4, Philip Hugenholtz2, Nikos C Kyrpides2, and Hans-Peter Klenk1* 1 DSMZ - German Collection of Microorganisms and Cell Cultures GmbH, Braunschweig, Germany 2 DOE Joint Genome Institute, Walnut Creek, California, USA 3 Los Alamos National Laboratory, Bioscience Division, Los Alamos, New Mexico, USA 4 Biological Data Management and Technology Center, Lawrence Berkeley National Laboratory, Berkeley, California, USA 5 Oak Ridge National Laboratory, Oak Ridge, Tennessee, USA 6 University of Regensburg, Microbiology – Archaeenzentrum, Regensburg, Germany 7 HZI – Helmholtz Centre for Infection Research, Braunschweig, Germany 8 University of California Davis Genome Center, Davis, California, USA *Corresponding author: Hans-Peter Klenk Keywords: hyperthermophilic, marine, strictly anaerobic, sulfate respiration, hydrogen utili- zation, hydrothermal systems, Archaeoglobaceae, GEBA Archaeoglobus profundus (Burggraf et al. 1990) is a hyperthermophilic archaeon in the eu- ryarchaeal class Archaeoglobi, which is currently represented by the single family Archaeog- lobaceae, containing six validly named species and two strains ascribed to the genus 'Geoglobus' which is taxonomically challenged as the corresponding type species has no va- lidly published name. -

Research Article Diversity and Distribution of Archaea in the Mangrove Sediment of Sundarbans
Hindawi Publishing Corporation Archaea Volume 2015, Article ID 968582, 14 pages http://dx.doi.org/10.1155/2015/968582 Research Article Diversity and Distribution of Archaea in the Mangrove Sediment of Sundarbans Anish Bhattacharyya,1 Niladri Shekhar Majumder,2 Pijush Basak,1 Shayantan Mukherji,3 Debojyoti Roy,1 Sudip Nag,1 Anwesha Haldar,4 Dhrubajyoti Chattopadhyay,1 Suparna Mitra,5 Maitree Bhattacharyya,1 and Abhrajyoti Ghosh3 1 Department of Biochemistry, University of Calcutta, 35 Ballygunge Circular Road, Kolkata, West Bengal 700019, India 2RocheDiagnosticsIndiaPvt.Ltd.,Block4C,AkashTower,NearRubyHospital,781Anandapur,Kolkata700107,India 3Department of Biochemistry, Bose Institute, P1/12, C. I. T. Road, Scheme VIIM, Kolkata, West Bengal 700054, India 4Department of Geography, University of Calcutta, 35 Ballygunge Circular Road, Kolkata, West Bengal 700019, India 5Norwich Medical School, University of East Anglia and Institute of Food Research, Norwich Research Park, Norwich, Norfolk NR4 7UA, UK Correspondence should be addressed to Dhrubajyoti Chattopadhyay; [email protected], Suparna Mitra; [email protected], Maitree Bhattacharyya; [email protected], and Abhrajyoti Ghosh; [email protected] Received 30 March 2015; Revised 25 June 2015; Accepted 14 July 2015 Academic Editor: William B. Whitman Copyright © 2015 Anish Bhattacharyya et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Mangroves are among the most diverse and productive coastal ecosystems in the tropical and subtropical regions. Environmental conditions particular to this biome make mangroves hotspots for microbial diversity, and the resident microbial communities play essential roles in maintenance of the ecosystem. -

Insights Into Archaeal Evolution and Symbiosis from the Genomes of a Nanoarchaeon and Its Inferred Crenarchaeal Host from Obsidian Pool, Yellowstone National Park
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Microbiology Publications and Other Works Microbiology 4-22-2013 Insights into archaeal evolution and symbiosis from the genomes of a nanoarchaeon and its inferred crenarchaeal host from Obsidian Pool, Yellowstone National Park Mircea Podar University of Tennessee - Knoxville, [email protected] Kira S. Makarova National Institutes of Health David E. Graham University of Tennessee - Knoxville, [email protected] Yuri I. Wolf National Institutes of Health Eugene V. Koonin National Institutes of Health See next page for additional authors Follow this and additional works at: https://trace.tennessee.edu/utk_micrpubs Part of the Microbiology Commons Recommended Citation Biology Direct 2013, 8:9 doi:10.1186/1745-6150-8-9 This Article is brought to you for free and open access by the Microbiology at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Microbiology Publications and Other Works by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. Authors Mircea Podar, Kira S. Makarova, David E. Graham, Yuri I. Wolf, Eugene V. Koonin, and Anna-Louise Reysenbach This article is available at TRACE: Tennessee Research and Creative Exchange: https://trace.tennessee.edu/ utk_micrpubs/44 Podar et al. Biology Direct 2013, 8:9 http://www.biology-direct.com/content/8/1/9 RESEARCH Open Access Insights into archaeal evolution and symbiosis from the genomes of a nanoarchaeon and its inferred crenarchaeal host from Obsidian Pool, Yellowstone National Park Mircea Podar1,2*, Kira S Makarova3, David E Graham1,2, Yuri I Wolf3, Eugene V Koonin3 and Anna-Louise Reysenbach4 Abstract Background: A single cultured marine organism, Nanoarchaeum equitans, represents the Nanoarchaeota branch of symbiotic Archaea, with a highly reduced genome and unusual features such as multiple split genes. -
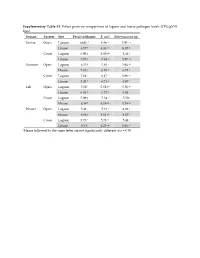
Supplementary Table S1. Fisher Pairwise Comparisons of Lagoon and House Pathogen Levels (CFU/Gvss Log10) Season System Site Fecal Coliforms E
Supplementary Table S1. Fisher pairwise comparisons of lagoon and house pathogen levels (CFU/gVSS log10) Season System Site Fecal coliforms E. coli Enterococcus sp. Spring Open Lagoon 6.00 g1 5.46 hi 5.91 cd House 6.57 e 6.33 cd 6.35 b Cover Lagoon 6.00 g 5.60 gh 5.44 f House 7.73 a 7.63 a 5.87 cd Summer Open Lagoon 6.35 f 5.83 f 5.94 cd House 7.83 a 6.10 e 6.73 a Cover Lagoon 7.04 c 6.47 c 5.86 cd House 7.42 b 6.71 b 6.07 c Fall Open Lagoon 5.58 i 5.58 gh 5.56 ef House 6.81 d 6.72 b 6.01 c Cover Lagoon 5.99 g 5.34 i 5.50 f House 6.38 f 6.19 de 5.74 de Winter Open Lagoon 5.41 j 5.13 j 4.88 g House 6.06 g 5.61 gh 6.67 a Cover Lagoon 5.73 h 5.73 fg 5.44 f House 6.34 f 6.25 de 5.86 cd 1Means followed by the same letter are not significantly different at p = 0.05 Supplementary Table S2. Relative abundances of OTUs identified using universal bacterial primer set, presented as relative abundances (%) Only bacterial families are counted in Figure 5 and discussion involving bacterial family identification. # Identified in # Identified from all 8 All 16 All #OTU Table SpOLRA SuOLRA FOLRA WOLRA SpCLRA SuCLRA FCLRA WCLRA SpOHRA SuOHRA FOHRA WOHRA SpCHRA SuCHRA FCHRA WCHRA all samples lagoon samples Samples Lagoons Notes Key Patulibacteraceae 0 9.93739E‐05* 0.000134 0.000116 0 0 0 0 0 0 0.000452 0 0 0 0 0 4 3 Sp = Spring O = Open Ruaniaceae 0 0 0 0 0 0 0 0 0 0 0 0 0 0.000221577 0 7.73E‐05 2 0 Su = Summer C = Covered Thermoactinomycetaceae 0 0 0.000267 0.000116 0 0.00037092 0.000529 0.000526 0 0.000342922 0.000271 0.000197 0 0 0 0 8 5 F = Fall L = Lagoon Beutenbergiaceae 0.000226334 0.000331246 0.000579 0.001663 0.000620176 0 0.000106 0.000807 0.002320743 0.000571537 0.000723 0.000591 0.000169731 0 0.000606 0 13 7 W = Winter H = House Thermoanaerobacterales Family III. -

The Genome Sequence of Methanohalophilus Mahii SLPT
Hindawi Publishing Corporation Archaea Volume 2010, Article ID 690737, 16 pages doi:10.1155/2010/690737 Research Article TheGenomeSequenceofMethanohalophilus mahii SLPT Reveals Differences in the Energy Metabolism among Members of the Methanosarcinaceae Inhabiting Freshwater and Saline Environments Stefan Spring,1 Carmen Scheuner,1 Alla Lapidus,2 Susan Lucas,2 Tijana Glavina Del Rio,2 Hope Tice,2 Alex Copeland,2 Jan-Fang Cheng,2 Feng Chen,2 Matt Nolan,2 Elizabeth Saunders,2, 3 Sam Pitluck,2 Konstantinos Liolios,2 Natalia Ivanova,2 Konstantinos Mavromatis,2 Athanasios Lykidis,2 Amrita Pati,2 Amy Chen,4 Krishna Palaniappan,4 Miriam Land,2, 5 Loren Hauser,2, 5 Yun-Juan Chang,2, 5 Cynthia D. Jeffries,2, 5 Lynne Goodwin,2, 3 John C. Detter,3 Thomas Brettin,3 Manfred Rohde,6 Markus Goker,¨ 1 Tanja Woyke, 2 Jim Bristow,2 Jonathan A. Eisen,2, 7 Victor Markowitz,4 Philip Hugenholtz,2 Nikos C. Kyrpides,2 and Hans-Peter Klenk1 1 DSMZ—German Collection of Microorganisms and Cell Cultures GmbH, 38124 Braunschweig, Germany 2 DOE Joint Genome Institute, Walnut Creek, CA 94598-1632, USA 3 Los Alamos National Laboratory, Bioscience Division, Los Alamos, NM 87545-001, USA 4 Biological Data Management and Technology Center, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA 5 Oak Ridge National Laboratory, Oak Ridge, TN 37830-8026, USA 6 HZI—Helmholtz Centre for Infection Research, 38124 Braunschweig, Germany 7 Davis Genome Center, University of California, Davis, CA 95817, USA Correspondence should be addressed to Stefan Spring, [email protected] and Hans-Peter Klenk, [email protected] Received 24 August 2010; Accepted 9 November 2010 Academic Editor: Valerie´ de Crecy-Lagard´ Copyright © 2010 Stefan Spring et al. -

Variations in the Two Last Steps of the Purine Biosynthetic Pathway in Prokaryotes
GBE Different Ways of Doing the Same: Variations in the Two Last Steps of the Purine Biosynthetic Pathway in Prokaryotes Dennifier Costa Brandao~ Cruz1, Lenon Lima Santana1, Alexandre Siqueira Guedes2, Jorge Teodoro de Souza3,*, and Phellippe Arthur Santos Marbach1,* 1CCAAB, Biological Sciences, Recoˆ ncavo da Bahia Federal University, Cruz das Almas, Bahia, Brazil 2Agronomy School, Federal University of Goias, Goiania,^ Goias, Brazil 3 Department of Phytopathology, Federal University of Lavras, Minas Gerais, Brazil Downloaded from https://academic.oup.com/gbe/article/11/4/1235/5345563 by guest on 27 September 2021 *Corresponding authors: E-mails: [email protected]fla.br; [email protected]. Accepted: February 16, 2019 Abstract The last two steps of the purine biosynthetic pathway may be catalyzed by different enzymes in prokaryotes. The genes that encode these enzymes include homologs of purH, purP, purO and those encoding the AICARFT and IMPCH domains of PurH, here named purV and purJ, respectively. In Bacteria, these reactions are mainly catalyzed by the domains AICARFT and IMPCH of PurH. In Archaea, these reactions may be carried out by PurH and also by PurP and PurO, both considered signatures of this domain and analogous to the AICARFT and IMPCH domains of PurH, respectively. These genes were searched for in 1,403 completely sequenced prokaryotic genomes publicly available. Our analyses revealed taxonomic patterns for the distribution of these genes and anticorrelations in their occurrence. The analyses of bacterial genomes revealed the existence of genes coding for PurV, PurJ, and PurO, which may no longer be considered signatures of the domain Archaea. Although highly divergent, the PurOs of Archaea and Bacteria show a high level of conservation in the amino acids of the active sites of the protein, allowing us to infer that these enzymes are analogs. -

Methanogens: Biochemical Background and Biotechnological Applications Franziska Enzmann1, Florian Mayer1 , Michael Rother2 and Dirk Holtmann1*
Enzmann et al. AMB Expr (2018) 8:1 https://doi.org/10.1186/s13568-017-0531-x MINI-REVIEW Open Access Methanogens: biochemical background and biotechnological applications Franziska Enzmann1, Florian Mayer1 , Michael Rother2 and Dirk Holtmann1* Abstract Since fossil sources for fuel and platform chemicals will become limited in the near future, it is important to develop new concepts for energy supply and production of basic reagents for chemical industry. One alternative to crude oil and fossil natural gas could be the biological conversion of CO2 or small organic molecules to methane via metha‑ nogenic archaea. This process has been known from biogas plants, but recently, new insights into the methanogenic metabolism, technical optimizations and new technology combinations were gained, which would allow moving beyond the mere conversion of biomass. In biogas plants, steps have been undertaken to increase yield and purity of the biogas, such as addition of hydrogen or metal granulate. Furthermore, the integration of electrodes led to the development of microbial electrosynthesis (MES). The idea behind this technique is to use CO 2 and electrical power to generate methane via the microbial metabolism. This review summarizes the biochemical and metabolic background of methanogenesis as well as the latest technical applications of methanogens. As a result, it shall give a sufcient overview over the topic to both, biologists and engineers handling biological or bioelectrochemical methanogenesis. Keywords: Methanogens, Genetic tools, Biogas, Microbial electrosynthesis, Electroactivity Introduction process involving methanogens. Tese archaea use CO 2 Methanogens are biocatalysts, which have the potential and H2 and/or small organic molecules, such as acetate, to contribute to a solution for future energy problems by formate, and methylamine and convert it to methane. -

Electrical Current Generation in Microbial Electrolysis Cells by Hyperthermophilic Archaea Ferroglobus Placidus and Geoglobus Ahangari
Bioelectrochemistry 119 (2018) 142–149 Contents lists available at ScienceDirect Bioelectrochemistry journal homepage: www.elsevier.com/locate/bioelechem Electrical current generation in microbial electrolysis cells by hyperthermophilic archaea Ferroglobus placidus and Geoglobus ahangari Yasemin D. Yilmazel a,b,⁎, Xiuping Zhu b,c,Kyoung-YeolKimb,DawnE.Holmesd, Bruce E. Logan b a Department of Chemical Engineering, Rochester Institute of Technology, Rochester, NY, USA b Department of Civil and Environmental Engineering, The Pennsylvania State University, University Park, PA, USA c Department of Civil and Environmental Engineering, Louisiana State University, Baton Rouge, LA, USA d Department of Biology, Western New England University, Springfield, MA, USA article info abstract Article history: Few microorganisms have been examined for current generation under thermophilic (40–65 °C) or hyperther- Received 5 May 2017 mophilic temperatures (≥80 °C) in microbial electrochemical systems. Two iron-reducing archaea from the fam- Received in revised form 27 September 2017 ily Archaeoglobaceae, Ferroglobus placidus and Geoglobus ahangari, showed electro-active behavior leading to Accepted 28 September 2017 current generation at hyperthermophilic temperatures in single-chamber microbial electrolysis cells (MECs). A Available online 02 October 2017 current density (j) of 0.68 ± 0.11 A/m2 was attained in F. placidus MECs at 85 °C, and 0.57 ± 0.10 A/m2 in G. ahangari MECs at 80 °C, with an applied voltage of 0.7 V. Cyclic voltammetry (CV) showed that both strains pro- Keywords: − − Hyperthermophilic archaea duced a sigmoidal catalytic wave, with a mid-point potential of 0.39 V (vs. Ag/AgCl) for F. placidus and 0.37 V Ferroglobus placidus for G. -

International Code of Nomenclature of Prokaryotes
2019, volume 69, issue 1A, pages S1–S111 International Code of Nomenclature of Prokaryotes Prokaryotic Code (2008 Revision) Charles T. Parker1, Brian J. Tindall2 and George M. Garrity3 (Editors) 1NamesforLife, LLC (East Lansing, Michigan, United States) 2Leibniz-Institut DSMZ-Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (Braunschweig, Germany) 3Michigan State University (East Lansing, Michigan, United States) Corresponding Author: George M. Garrity ([email protected]) Table of Contents 1. Foreword to the First Edition S1–S1 2. Preface to the First Edition S2–S2 3. Preface to the 1975 Edition S3–S4 4. Preface to the 1990 Edition S5–S6 5. Preface to the Current Edition S7–S8 6. Memorial to Professor R. E. Buchanan S9–S12 7. Chapter 1. General Considerations S13–S14 8. Chapter 2. Principles S15–S16 9. Chapter 3. Rules of Nomenclature with Recommendations S17–S40 10. Chapter 4. Advisory Notes S41–S42 11. References S43–S44 12. Appendix 1. Codes of Nomenclature S45–S48 13. Appendix 2. Approved Lists of Bacterial Names S49–S49 14. Appendix 3. Published Sources for Names of Prokaryotic, Algal, Protozoal, Fungal, and Viral Taxa S50–S51 15. Appendix 4. Conserved and Rejected Names of Prokaryotic Taxa S52–S57 16. Appendix 5. Opinions Relating to the Nomenclature of Prokaryotes S58–S77 17. Appendix 6. Published Sources for Recommended Minimal Descriptions S78–S78 18. Appendix 7. Publication of a New Name S79–S80 19. Appendix 8. Preparation of a Request for an Opinion S81–S81 20. Appendix 9. Orthography S82–S89 21. Appendix 10. Infrasubspecific Subdivisions S90–S91 22. Appendix 11. The Provisional Status of Candidatus S92–S93 23. -

A Metagenomics Roadmap to the Uncultured Genome Diversity in Hypersaline Soda Lake Sediments Charlotte D
Vavourakis et al. Microbiome (2018) 6:168 https://doi.org/10.1186/s40168-018-0548-7 RESEARCH Open Access A metagenomics roadmap to the uncultured genome diversity in hypersaline soda lake sediments Charlotte D. Vavourakis1 , Adrian-Stefan Andrei2†, Maliheh Mehrshad2†, Rohit Ghai2, Dimitry Y. Sorokin3,4 and Gerard Muyzer1* Abstract Background: Hypersaline soda lakes are characterized by extreme high soluble carbonate alkalinity. Despite the high pH and salt content, highly diverse microbial communities are known to be present in soda lake brines but the microbiome of soda lake sediments received much less attention of microbiologists. Here, we performed metagenomic sequencing on soda lake sediments to give the first extensive overview of the taxonomic diversity found in these complex, extreme environments and to gain novel physiological insights into the most abundant, uncultured prokaryote lineages. Results: We sequenced five metagenomes obtained from four surface sediments of Siberian soda lakes with a pH 10 and a salt content between 70 and 400 g L−1. The recovered 16S rRNA gene sequences were mostly from Bacteria,evenin the salt-saturated lakes. Most OTUs were assigned to uncultured families. We reconstructed 871 metagenome-assembled genomes (MAGs) spanning more than 45 phyla and discovered the first extremophilic members of the Candidate Phyla Radiation (CPR). Five new species of CPR were among the most dominant community members. Novel dominant lineages were found within previously well-characterized functional groups involved in carbon, sulfur, and nitrogen cycling. Moreover, key enzymes of the Wood-Ljungdahl pathway were encoded within at least four bacterial phyla never previously associated with this ancient anaerobic pathway for carbon fixation and dissimilation, including the Actinobacteria.