Archaeoglobus Profundus Type Strain (AV18T)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genome-Resolved Meta-Analysis of the Microbiome in Oil Reservoirs Worldwide
microorganisms Article Genome-Resolved Meta-Analysis of the Microbiome in Oil Reservoirs Worldwide Kelly J. Hidalgo 1,2,* , Isabel N. Sierra-Garcia 3 , German Zafra 4 and Valéria M. de Oliveira 1 1 Microbial Resources Division, Research Center for Chemistry, Biology and Agriculture (CPQBA), University of Campinas–UNICAMP, Av. Alexandre Cazellato 999, 13148-218 Paulínia, Brazil; [email protected] 2 Graduate Program in Genetics and Molecular Biology, Institute of Biology, University of Campinas (UNICAMP), Rua Monteiro Lobato 255, Cidade Universitária, 13083-862 Campinas, Brazil 3 Biology Department & CESAM, University of Aveiro, Aveiro, Portugal, Campus de Santiago, Avenida João Jacinto de Magalhães, 3810-193 Aveiro, Portugal; [email protected] 4 Grupo de Investigación en Bioquímica y Microbiología (GIBIM), Escuela de Microbiología, Universidad Industrial de Santander, Cra 27 calle 9, 680002 Bucaramanga, Colombia; [email protected] * Correspondence: [email protected]; Tel.: +55-19981721510 Abstract: Microorganisms inhabiting subsurface petroleum reservoirs are key players in biochemical transformations. The interactions of microbial communities in these environments are highly complex and still poorly understood. This work aimed to assess publicly available metagenomes from oil reservoirs and implement a robust pipeline of genome-resolved metagenomics to decipher metabolic and taxonomic profiles of petroleum reservoirs worldwide. Analysis of 301.2 Gb of metagenomic information derived from heavily flooded petroleum reservoirs in China and Alaska to non-flooded petroleum reservoirs in Brazil enabled us to reconstruct 148 metagenome-assembled genomes (MAGs) of high and medium quality. At the phylum level, 74% of MAGs belonged to bacteria and 26% to archaea. The profiles of these MAGs were related to the physicochemical parameters and recovery management applied. -

A Web Tool for Hydrogenase Classification and Analysis Dan Søndergaard1, Christian N
www.nature.com/scientificreports OPEN HydDB: A web tool for hydrogenase classification and analysis Dan Søndergaard1, Christian N. S. Pedersen1 & Chris Greening2,3 H2 metabolism is proposed to be the most ancient and diverse mechanism of energy-conservation. The Received: 24 June 2016 metalloenzymes mediating this metabolism, hydrogenases, are encoded by over 60 microbial phyla Accepted: 09 September 2016 and are present in all major ecosystems. We developed a classification system and web tool, HydDB, Published: 27 September 2016 for the structural and functional analysis of these enzymes. We show that hydrogenase function can be predicted by primary sequence alone using an expanded classification scheme (comprising 29 [NiFe], 8 [FeFe], and 1 [Fe] hydrogenase classes) that defines 11 new classes with distinct biological functions. Using this scheme, we built a web tool that rapidly and reliably classifies hydrogenase primary sequences using a combination of k-nearest neighbors’ algorithms and CDD referencing. Demonstrating its capacity, the tool reliably predicted hydrogenase content and function in 12 newly-sequenced bacteria, archaea, and eukaryotes. HydDB provides the capacity to browse the amino acid sequences of 3248 annotated hydrogenase catalytic subunits and also contains a detailed repository of physiological, biochemical, and structural information about the 38 hydrogenase classes defined here. The database and classifier are freely and publicly available at http://services.birc.au.dk/hyddb/ Microorganisms conserve energy by metabolizing H2. Oxidation of this high-energy fuel yields electrons that can be used for respiration and carbon-fixation. This diffusible gas is also produced in diverse fermentation and 1 anaerobic respiratory processes . H2 metabolism contributes to the growth and survival of microorganisms across the three domains of life, including chemotrophs and phototrophs, lithotrophs and heterotrophs, aerobes and 1,2 anaerobes, mesophiles and extremophiles alike . -

Assessment of the Carbon Monoxide Metabolism of the Hyperthermophilic Sulfate-Reducing Archaeon Archaeoglobus Fulgidus VC-16 by Comparative Transcriptome Analyses
Hindawi Publishing Corporation Archaea Volume 2015, Article ID 235384, 12 pages http://dx.doi.org/10.1155/2015/235384 Research Article Assessment of the Carbon Monoxide Metabolism of the Hyperthermophilic Sulfate-Reducing Archaeon Archaeoglobus fulgidus VC-16 by Comparative Transcriptome Analyses William P. Hocking, Irene Roalkvam, Carina Magnussen, Runar Stokke, and Ida H. Steen Department of Biology, Centre for Geobiology, University of Bergen, 5020 Bergen, Norway Correspondence should be addressed to Ida H. Steen; [email protected] Received 10 April 2015; Revised 9 June 2015; Accepted 14 June 2015 Academic Editor: Uwe Deppenmeier Copyright © 2015 William P. Hocking et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The hyperthermophilic, sulfate-reducing archaeon, Archaeoglobus fulgidus, utilizes CO as an energy source and it is resistant to the toxic effects of high CO concentrations. Herein, transcription profiles were obtained from A. fulgidus during growth with CO and sulfate or thiosulfate, or without an electron acceptor. This provided a basis for a model of the CO metabolism of A. fulgidus. The model suggests proton translocation by “Mitchell-type” loops facilitated by Fqo catalyzingred aFd :menaquinone oxidoreductase reaction, as the major mode of energy conservation, rather than formate or H2 cycling during respiratory growth. The bifunctional CODH (cdhAB-2) is predicted to play an ubiquitous role in the metabolism of CO, and a novel nitrate reductase- associated respiratory complex was induced specifically in the presence of sulfate. A potential role of this complex in relation to Fdred and APS reduction is discussed. -

Specific Enrichment of Hyperthermophilic Electroactive Archaea From
bioRxiv preprint doi: https://doi.org/10.1101/272039; this version posted February 27, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Specific enrichment of hyperthermophilic electroactive Archaea from 2 deep-sea hydrothermal vent on electrically conductive support. 3 4 Guillaume Pillot1, Eléonore Frouin1, Emilie Pasero1, Anne Godfroy2, Yannick Combet-Blanc1, 5 Sylvain Davidson1 & Pierre-Pol Liebgott1* 6 1 Aix Marseille Université, IRD, Université de Toulon, CNRS, MIO UM 110, Marseille, France. 7 2 Laboratoire de Microbiologie des Environnements Extrêmes - UMR6197 IFREMER, CNRS, 8 UBO Centre de Brest–CS10070/IUEM, Plouzané, France. 9 *Corresponding author: [email protected]; Mediterranean Institute of 10 Oceanography, Campus de Luminy, Bâtiment OCEANOMED, 13288 Marseille Cedex 09. 11 Abstract 12 While more and more investigations are done to isolate hyperthermophilic exoelectrogenic 13 communities from environments, none have been performed yet on deep-sea hydrothermal vent. 14 Samples of black smoker chimney from Rainbow site on the Atlantic mid-oceanic ridge have 15 been harvested for enriching exoelectrogens in microbial electrolysis cells under hyperthermophilic 16 (80°C) condition. Two enrichments have been performed: one from direct inoculation of crushed 17 chimney and the other one from inoculation of a pre-cultivation on iron (III) oxide. In both 18 experiments, a current production was observed from 2.4 A/m² to 5.8 A/m² with a set anode 19 potential of +0.05 vs SHE. Taxonomic affiliation of the exoelectrogen communities obtained 20 exhibited a specific enrichment of Archaea from Thermococcales and Archeoglobales orders on the 21 electrode, even when both inocula were dominated by Bacteria. -

Microbial and Geochemical Investigation Down to 2000 M Deep Triassic Rock (Meuse/Haute Marne, France)
geosciences Article Microbial and Geochemical Investigation down to 2000 m Deep Triassic Rock (Meuse/Haute Marne, France) Vanessa Leblanc 1,2,3, Jennifer Hellal 1 , Marie-Laure Fardeau 4,5, Saber Khelaifia 4,5, Claire Sergeant 2,3, Francis Garrido 1, Bernard Ollivier 4,5 and Catherine Joulian 1,* 1 BRGM, Geomicrobiology and Environmental Monitoring Unit, F-45060 Orléans CEDEX 02, France; [email protected] (V.L.); [email protected] (J.H.); [email protected] (F.G.) 2 Bordeaux University, Centre d’Etudes Nucleaires de Bordeaux Gradignan, UMR5797, F-33170 Gradignan, France; [email protected] 3 CNRS-IN2P3, Centre d’Etudes Nucleaires de Bordeaux Gradignan, UMR5797, F-33170 Gradignan, France 4 Aix Marseille Université, CNRS/INSU, IRD, Mediterranean Institute of Oceanography (MIO), UM 110, 13288 Marseille, France; [email protected] (M.-L.F.); Saber.Khelaifi[email protected] (S.K.); [email protected] (B.O.) 5 Université de Toulon, CNRS/INSU, 83957 La Garde, France * Correspondence: [email protected]; Tel.: +33-2-3864-3089 Received: 31 August 2018; Accepted: 13 December 2018; Published: 20 December 2018 Abstract: In 2008, as part of a feasibility study for radioactive waste disposal in deep geological formations, the French National Radioactive Waste Management Agency (ANDRA) drilled several boreholes in the transposition zone in order to define the potential variations in the properties of the Callovo–Oxfordian claystone formation. This consisted of a rare opportunity to investigate the deep continental biosphere that is still poorly known. Four rock cores, from 1709, 1804, 1865, and 1935 m below land surface, were collected from Lower and Middle Triassic formations in the Paris Basin (France) to investigate their microbial and geochemical composition. -

Assessment of the Carbon Monoxide Metabolism of the Hyperthermophilic Sulfate-Reducing Archaeon Archaeoglobus Fulgidus VC-16 by Comparative Transcriptome Analyses
Hindawi Publishing Corporation Archaea Volume 2015, Article ID 235384, 12 pages http://dx.doi.org/10.1155/2015/235384 Research Article Assessment of the Carbon Monoxide Metabolism of the Hyperthermophilic Sulfate-Reducing Archaeon Archaeoglobus fulgidus VC-16 by Comparative Transcriptome Analyses William P. Hocking, Irene Roalkvam, Carina Magnussen, Runar Stokke, and Ida H. Steen Department of Biology, Centre for Geobiology, University of Bergen, 5020 Bergen, Norway Correspondence should be addressed to Ida H. Steen; [email protected] Received 10 April 2015; Revised 9 June 2015; Accepted 14 June 2015 Academic Editor: Uwe Deppenmeier Copyright © 2015 William P. Hocking et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The hyperthermophilic, sulfate-reducing archaeon, Archaeoglobus fulgidus, utilizes CO as an energy source and it is resistant to the toxic effects of high CO concentrations. Herein, transcription profiles were obtained from A. fulgidus during growth with CO and sulfate or thiosulfate, or without an electron acceptor. This provided a basis for a model of the CO metabolism of A. fulgidus. The model suggests proton translocation by “Mitchell-type” loops facilitated by Fqo catalyzingred aFd :menaquinone oxidoreductase reaction, as the major mode of energy conservation, rather than formate or H2 cycling during respiratory growth. The bifunctional CODH (cdhAB-2) is predicted to play an ubiquitous role in the metabolism of CO, and a novel nitrate reductase- associated respiratory complex was induced specifically in the presence of sulfate. A potential role of this complex in relation to Fdred and APS reduction is discussed. -
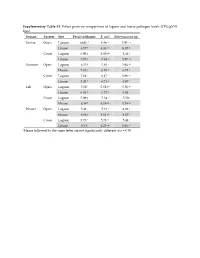
Supplementary Table S1. Fisher Pairwise Comparisons of Lagoon and House Pathogen Levels (CFU/Gvss Log10) Season System Site Fecal Coliforms E
Supplementary Table S1. Fisher pairwise comparisons of lagoon and house pathogen levels (CFU/gVSS log10) Season System Site Fecal coliforms E. coli Enterococcus sp. Spring Open Lagoon 6.00 g1 5.46 hi 5.91 cd House 6.57 e 6.33 cd 6.35 b Cover Lagoon 6.00 g 5.60 gh 5.44 f House 7.73 a 7.63 a 5.87 cd Summer Open Lagoon 6.35 f 5.83 f 5.94 cd House 7.83 a 6.10 e 6.73 a Cover Lagoon 7.04 c 6.47 c 5.86 cd House 7.42 b 6.71 b 6.07 c Fall Open Lagoon 5.58 i 5.58 gh 5.56 ef House 6.81 d 6.72 b 6.01 c Cover Lagoon 5.99 g 5.34 i 5.50 f House 6.38 f 6.19 de 5.74 de Winter Open Lagoon 5.41 j 5.13 j 4.88 g House 6.06 g 5.61 gh 6.67 a Cover Lagoon 5.73 h 5.73 fg 5.44 f House 6.34 f 6.25 de 5.86 cd 1Means followed by the same letter are not significantly different at p = 0.05 Supplementary Table S2. Relative abundances of OTUs identified using universal bacterial primer set, presented as relative abundances (%) Only bacterial families are counted in Figure 5 and discussion involving bacterial family identification. # Identified in # Identified from all 8 All 16 All #OTU Table SpOLRA SuOLRA FOLRA WOLRA SpCLRA SuCLRA FCLRA WCLRA SpOHRA SuOHRA FOHRA WOHRA SpCHRA SuCHRA FCHRA WCHRA all samples lagoon samples Samples Lagoons Notes Key Patulibacteraceae 0 9.93739E‐05* 0.000134 0.000116 0 0 0 0 0 0 0.000452 0 0 0 0 0 4 3 Sp = Spring O = Open Ruaniaceae 0 0 0 0 0 0 0 0 0 0 0 0 0 0.000221577 0 7.73E‐05 2 0 Su = Summer C = Covered Thermoactinomycetaceae 0 0 0.000267 0.000116 0 0.00037092 0.000529 0.000526 0 0.000342922 0.000271 0.000197 0 0 0 0 8 5 F = Fall L = Lagoon Beutenbergiaceae 0.000226334 0.000331246 0.000579 0.001663 0.000620176 0 0.000106 0.000807 0.002320743 0.000571537 0.000723 0.000591 0.000169731 0 0.000606 0 13 7 W = Winter H = House Thermoanaerobacterales Family III. -

Electrical Current Generation in Microbial Electrolysis Cells by Hyperthermophilic Archaea Ferroglobus Placidus and Geoglobus Ahangari
Bioelectrochemistry 119 (2018) 142–149 Contents lists available at ScienceDirect Bioelectrochemistry journal homepage: www.elsevier.com/locate/bioelechem Electrical current generation in microbial electrolysis cells by hyperthermophilic archaea Ferroglobus placidus and Geoglobus ahangari Yasemin D. Yilmazel a,b,⁎, Xiuping Zhu b,c,Kyoung-YeolKimb,DawnE.Holmesd, Bruce E. Logan b a Department of Chemical Engineering, Rochester Institute of Technology, Rochester, NY, USA b Department of Civil and Environmental Engineering, The Pennsylvania State University, University Park, PA, USA c Department of Civil and Environmental Engineering, Louisiana State University, Baton Rouge, LA, USA d Department of Biology, Western New England University, Springfield, MA, USA article info abstract Article history: Few microorganisms have been examined for current generation under thermophilic (40–65 °C) or hyperther- Received 5 May 2017 mophilic temperatures (≥80 °C) in microbial electrochemical systems. Two iron-reducing archaea from the fam- Received in revised form 27 September 2017 ily Archaeoglobaceae, Ferroglobus placidus and Geoglobus ahangari, showed electro-active behavior leading to Accepted 28 September 2017 current generation at hyperthermophilic temperatures in single-chamber microbial electrolysis cells (MECs). A Available online 02 October 2017 current density (j) of 0.68 ± 0.11 A/m2 was attained in F. placidus MECs at 85 °C, and 0.57 ± 0.10 A/m2 in G. ahangari MECs at 80 °C, with an applied voltage of 0.7 V. Cyclic voltammetry (CV) showed that both strains pro- Keywords: − − Hyperthermophilic archaea duced a sigmoidal catalytic wave, with a mid-point potential of 0.39 V (vs. Ag/AgCl) for F. placidus and 0.37 V Ferroglobus placidus for G. -

Divergent Methyl-Coenzyme M Reductase Genes in a Deep-Subseafloor Archaeoglobi
The ISME Journal (2019) 13:1269–1279 https://doi.org/10.1038/s41396-018-0343-2 ARTICLE Divergent methyl-coenzyme M reductase genes in a deep-subseafloor Archaeoglobi 1 2,3 1 1 1 4 Joel A. Boyd ● Sean P. Jungbluth ● Andy O. Leu ● Paul N. Evans ● Ben J. Woodcroft ● Grayson L. Chadwick ● 4 5 6 1 Victoria J. Orphan ● Jan P. Amend ● Michael S. Rappé ● Gene W. Tyson Received: 7 October 2018 / Revised: 29 November 2018 / Accepted: 11 December 2018 / Published online: 16 January 2019 © The Author(s) 2019. This article is published with open access Abstract The methyl-coenzyme M reductase (MCR) complex is a key enzyme in archaeal methane generation and has recently been proposed to also be involved in the oxidation of short-chain hydrocarbons including methane, butane, and potentially propane. The number of archaeal clades encoding the MCR continues to grow, suggesting that this complex was inherited from an ancient ancestor, or has undergone extensive horizontal gene transfer. Expanding the representation of MCR- encoding lineages through metagenomic approaches will help resolve the evolutionary history of this complex. Here, a near- complete Archaeoglobi metagenome-assembled genome (MAG; Ca. Polytropus marinifundus gen. nov. sp. nov.) was fl fl 1234567890();,: 1234567890();,: recovered from the deep subsea oor along the Juan de Fuca Ridge ank that encodes two divergent McrABG operons similar to those found in Ca. Bathyarchaeota and Ca. Syntrophoarchaeum MAGs. Ca. P. marinifundus is basal to members of the class Archaeoglobi, and encodes the genes for β-oxidation, potentially allowing an alkanotrophic metabolism similar to that proposed for Ca. -

University of Florida Thesis Or Dissertation Formatting
CHARACTERIZING THE MOLECULAR MECHANISMS CONTRIBUTING TO BIOLOGICALLY INDUCED CARBONATE MINERALIZATION AND THROMBOLITE FORMATION By ARTEMIS S. LOUYAKIS A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY UNIVERSITY OF FLORIDA 2017 © 2017 Artemis S. Louyakis To my mother, for supporting every single goal I’ve ever had, the memory of my father, for keeping me focused, and my partner, for all he’s done ACKNOWLEDGMENTS I would like to begin by acknowledging and thanking my mentor, Dr. Jamie Foster, for all her guidance throughout this Ph.D. I thank my committee members for all of their advice and support - Drs. Eric Triplett, Julie Maupin, Nian Wang, and Eric McLamore. I’d like to thank the rest of the Department of Microbiology and Cell Science, staff for always keeping my academic life in order, faculty for never turning me away when I came to use equipment or ask for help, especially Drs. K.T. Shanmugan and Wayne Nicholson, as well as Dr. Andy Schuerger from the Dept. of Plant Pathology for his advice over the years. I’d also like to acknowledge those lab members and extended lab members who made themselves readily available to talk through any problems I came up against and celebrate when all went well, including Drs. Rafael Oliveira, Jennifer Mobberley, and Giorgio Casaburi, and Lexi Duscher, Rachelle Banjawo, Maddie Vroom, Hadrien Gourlé, and so many more. I’d also like to profusely thank my family and friends who have never been anything less than completely supportive of me, specifically my partner Nathan Prince, my mother and siblings Denise Louyakis, Bobbi Louyakis, Nick Newman, Cori Sergi, extended parents and siblings Carol Prince, Barry Prince, Aaron Prince, my nieces and nephew Bailey O’Regan, Bella O’Regan, Layla Newman, Colton Prince, and Summer Prince, and my dearest friends Tina Pontbriand, Tom Pontbriand, Karen Chan, Dalal Haouchar, Alexi Casaburi, and Eloise Stikeman. -

Phylogeny Based on 16S Rrna/DNA
Phylogeny Based on Secondary article 16S rRNA/DNA Article Contents . Introduction Erko Stackebrandt, DSMZ-German Collection of Microorganisms and Cell Cultures GmbH, . Semantic Macromolecules: a Basis for Phylogenetic Braunschweig, Germany Studies . Sequence Determination, Sequence Alignment and Determination of Sequence Similarities Modern systematics of prokaryotes is based on comparative analysis of the evolutionarily . Recognition of the Higher Taxa of Prokaryotes conservative genes coding for 16S ribosomal RNA. Dendrograms of phylogenetic . Polyphasic Approach to Bacterial Systematics relatedness show the order in which organisms evolved in time, thus providing a basis for . The Taxonomic Rank ‘Species’ in Bacteriology their classification. Introduction are the historical record of evolution, and the determina- In contrast to more highly evolved eukaryotes, in which tion of their primary structure provides a powerful means complex morphologies visibly reflect their evolutionary by which evolutionary relationships can be measured. In history, the microscopic and ultrastructural features of essence, two organisms possessing a given stretch of microorganisms cannot be used to deduce the way in which semantides which differ in only a few changes (mutations, the prokaryotes and the morphologically simple eukar- nucleotide order or amino acid positions) are more closely yotic forms evolved. Before 1960 taxonomists were unable related to each other than those organisms in which a to appreciate the complexity of microbial systematics and higher number of changes have accumulated. Thus these to recognize that groups based on superficial properties molecules can be considered as chronometers. As different alone did not necessarily reflect those which arose due to genes are subjected to different rates of changes (same evolutionary processes. -

Systematic Bacteriology Second Edition
BERGEY'S MANUAL® OF Systematic Bacteriology Second Edition Volume One The Archaea and the Deeply Branching and Phototrophic Bacteria Springer New York Berlin Heidelberg Barcelona HongKong London Milan Paris Singapore Tokyo BERGEY'S MANUAL® OF Systematic Bacteriology Second Edition Volume One The Archaea and the Deeply Branching and Phototrophic Bacteria David R. Boone Richard W. Castenholz EDITORS, VOLUME ONE George M. Garrity EDITOR-IN-CHIEF EDITORIAL BOARD James T. Staley, Chairman, David R. Boone, Vice Chairman, Don J. Brenner, Richard W. Castenholz, George M. Garrity, Michael Goodfellow, Noel R. Krieg, Fred A. Rainey, Karl-Heinz Schleifer WITH CONTRIBUTIONS FROM 105 COLLEAGUES Springer George M. Garrity Department of Microbiology and Molecular Genetics Bergey's Manual Trust Michigan State University East Lansing, MI 48824-1101 USA Library of Congress Cataloging-in-Publication Data Bergey's manual of systematic bacteriology / David R. Boone, Richard W. Castenholz, editors, volume 1 ; George M. Garrity, editor-in-chief.-2nd ed. p.cm. Includes bibliographical references and index. Contents: v.I. The archaea and the deeply branching and phototrophic bacteria. ISBN 0-387-98771-1 (alk. paper) 1. Bacteria-Classification. I. Title: Systematic bacteriology. II. Boone, David R. III. Castenholz, Richard W. IV. Garrity, George M. QR81.B462001 579.3'01 '2-dc21 2001020400 With 330 illustrations The following proprietary names of products are used in this volume: Casamino@ acids; Vector NTI®; XLI0-Gold®; Gelrite®; Tryptone@l; Phytagel@; bio-Trypcasew; Trypticasev; Oxoid® purified agar. Printed on acid-free paper. First edition published 1984-1989 by Bergey's Manual Trust and Williams & Wilkins, Baltimore. © 2001 Bergey's Manual Trust All rights reserved.