Rabbit Anti-SNAPC3/FITC Conjugated Antibody
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PARSANA-DISSERTATION-2020.Pdf
DECIPHERING TRANSCRIPTIONAL PATTERNS OF GENE REGULATION: A COMPUTATIONAL APPROACH by Princy Parsana A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2020 © 2020 Princy Parsana All rights reserved Abstract With rapid advancements in sequencing technology, we now have the ability to sequence the entire human genome, and to quantify expression of tens of thousands of genes from hundreds of individuals. This provides an extraordinary opportunity to learn phenotype relevant genomic patterns that can improve our understanding of molecular and cellular processes underlying a trait. The high dimensional nature of genomic data presents a range of computational and statistical challenges. This dissertation presents a compilation of projects that were driven by the motivation to efficiently capture gene regulatory patterns in the human transcriptome, while addressing statistical and computational challenges that accompany this data. We attempt to address two major difficulties in this domain: a) artifacts and noise in transcriptomic data, andb) limited statistical power. First, we present our work on investigating the effect of artifactual variation in gene expression data and its impact on trans-eQTL discovery. Here we performed an in-depth analysis of diverse pre-recorded covariates and latent confounders to understand their contribution to heterogeneity in gene expression measurements. Next, we discovered 673 trans-eQTLs across 16 human tissues using v6 data from the Genotype Tissue Expression (GTEx) project. Finally, we characterized two trait-associated trans-eQTLs; one in Skeletal Muscle and another in Thyroid. Second, we present a principal component based residualization method to correct gene expression measurements prior to reconstruction of co-expression networks. -
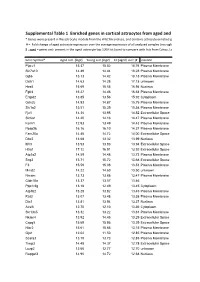
Supplemental Table 1 Enriched Genes in Cortical Astrocytes from Aged
Supplemental Table 1 Enriched genes in cortical astrocytes from aged and young-adult mice * Genes were present in the astrocyte module from the WGCNA analysis, and contains astrocyte enriched genes compared to microglia and oligodendrocytes # = Fold change of aged astrocyte expression over the average expression of all analyzed samples (microglia, astrocytes: young, old, with and without myelin contamination) $ ; aged = genes only present in the aged astrocyte top 1000 list (used to compare with lists from Cahoy, Lovatt, Doyle; see Fig. 4B), all = genes present in all astrocyte top 1000 lists Gene Symbol* Aged astr. (log2) Young astr.(log2) FC (aged/ aver.)# Location Ptprz1 15.37 15.02 18.76 Plasma Membrane Slc7a10 14.49 14.44 18.28 Plasma Membrane Gjb6 15.13 14.42 18.18 Plasma Membrane Dclk1 14.63 14.28 17.18 unknown Hes5 15.69 15.55 16.94 Nucleus Fgfr3 15.27 14.46 16.54 Plasma Membrane Entpd2 13.85 13.56 15.92 Cytoplasm Grin2c 14.93 14.87 15.75 Plasma Membrane Slc1a2 15.51 15.39 15.58 Plasma Membrane Fjx1 14.36 13.98 14.52 Extracellular Space Slc6a1 14.20 14.16 14.47 Plasma Membrane Kcnk1 12.93 13.49 14.43 Plasma Membrane Ppap2b 16.16 16.10 14.37 Plasma Membrane Fam20a 14.48 14.72 14.00 Extracellular Space Dbx2 13.68 13.32 13.99 Nucleus Itih3 13.93 13.93 13.94 Extracellular Space Htra1 17.12 16.91 13.92 Extracellular Space Atp1a2 14.59 14.48 13.73 Plasma Membrane Scg3 15.71 15.72 13.68 Extracellular Space F3 15.59 15.08 13.51 Plasma Membrane Mmd2 14.22 14.60 13.50 unknown Nrcam 13.73 13.88 13.47 Plasma Membrane Cldn10a 13.37 13.57 13.46 -

New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology
Cancer Research and Treatment 2003;35(4):304-313 New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology Yong-Wan Kim, Ph.D.1, Min-Je Suh, M.S.1, Jin-Sik Bae, M.S.1, Su Mi Bae, M.S.1, Joo Hee Yoon, M.D.2, Soo Young Hur, M.D.2, Jae Hoon Kim, M.D.2, Duck Young Ro, M.D.2, Joon Mo Lee, M.D.2, Sung Eun Namkoong, M.D.2, Chong Kook Kim, Ph.D.3 and Woong Shick Ahn, M.D.2 1Catholic Research Institutes of Medical Science, 2Department of Obstetrics and Gynecology, College of Medicine, The Catholic University of Korea, Seoul; 3College of Pharmacy, Seoul National University, Seoul, Korea Purpose: This study utilized both mRNA differential significant genes of unknown function affected by the display and the Gene Ontology (GO) analysis to char- HPV-16-derived pathway. The GO analysis suggested that acterize the multiple interactions of a number of genes the cervical cancer cells underwent repression of the with gene expression profiles involved in the HPV-16- cancer-specific cell adhesive properties. Also, genes induced cervical carcinogenesis. belonging to DNA metabolism, such as DNA repair and Materials and Methods: mRNA differential displays, replication, were strongly down-regulated, whereas sig- with HPV-16 positive cervical cancer cell line (SiHa), and nificant increases were shown in the protein degradation normal human keratinocyte cell line (HaCaT) as a con- and synthesis. trol, were used. Each human gene has several biological Conclusion: The GO analysis can overcome the com- functions in the Gene Ontology; therefore, several func- plexity of the gene expression profile of the HPV-16- tions of each gene were chosen to establish a powerful associated pathway, identify several cancer-specific cel- cervical carcinogenesis pathway. -

Genetic Basis of Simple and Complex Traits with Relevance to Avian Evolution
Genetic basis of simple and complex traits with relevance to avian evolution Małgorzata Anna Gazda Doctoral Program in Biodiversity, Genetics and Evolution D Faculdade de Ciências da Universidade do Porto 2019 Supervisor Miguel Jorge Pinto Carneiro, Auxiliary Researcher, CIBIO/InBIO, Laboratório Associado, Universidade do Porto Co-supervisor Ricardo Lopes, CIBIO/InBIO Leif Andersson, Uppsala University FCUP Genetic basis of avian traits Nota Previa Na elaboração desta tese, e nos termos do número 2 do Artigo 4º do Regulamento Geral dos Terceiros Ciclos de Estudos da Universidade do Porto e do Artigo 31º do D.L.74/2006, de 24 de Março, com a nova redação introduzida pelo D.L. 230/2009, de 14 de Setembro, foi efetuado o aproveitamento total de um conjunto coerente de trabalhos de investigação já publicados ou submetidos para publicação em revistas internacionais indexadas e com arbitragem científica, os quais integram alguns dos capítulos da presente tese. Tendo em conta que os referidos trabalhos foram realizados com a colaboração de outros autores, o candidato esclarece que, em todos eles, participou ativamente na sua conceção, na obtenção, análise e discussão de resultados, bem como na elaboração da sua forma publicada. Este trabalho foi apoiado pela Fundação para a Ciência e Tecnologia (FCT) através da atribuição de uma bolsa de doutoramento (PD/BD/114042/2015) no âmbito do programa doutoral em Biodiversidade, Genética e Evolução (BIODIV). 2 FCUP Genetic basis of avian traits Acknowledgements Firstly, I would like to thank to my all supervisors Miguel Carneiro, Ricardo Lopes and Leif Andersson, for the demanding task of supervising myself last four years. -

Distinguishing Pleiotropy from Linked QTL Between Milk Production Traits
Cai et al. Genet Sel Evol (2020) 52:19 https://doi.org/10.1186/s12711-020-00538-6 Genetics Selection Evolution RESEARCH ARTICLE Open Access Distinguishing pleiotropy from linked QTL between milk production traits and mastitis resistance in Nordic Holstein cattle Zexi Cai1*†, Magdalena Dusza2†, Bernt Guldbrandtsen1, Mogens Sandø Lund1 and Goutam Sahana1 Abstract Background: Production and health traits are central in cattle breeding. Advances in next-generation sequencing technologies and genotype imputation have increased the resolution of gene mapping based on genome-wide association studies (GWAS). Thus, numerous candidate genes that afect milk yield, milk composition, and mastitis resistance in dairy cattle are reported in the literature. Efect-bearing variants often afect multiple traits. Because the detection of overlapping quantitative trait loci (QTL) regions from single-trait GWAS is too inaccurate and subjective, multi-trait analysis is a better approach to detect pleiotropic efects of variants in candidate genes. However, large sample sizes are required to achieve sufcient power. Multi-trait meta-analysis is one approach to deal with this prob- lem. Thus, we performed two multi-trait meta-analyses, one for three milk production traits (milk yield, protein yield and fat yield), and one for milk yield and mastitis resistance. Results: For highly correlated traits, the power to detect pleiotropy was increased by multi-trait meta-analysis com- pared with the subjective assessment of overlapping of single-trait QTL confdence intervals. Pleiotropic efects of lead single nucleotide polymorphisms (SNPs) that were detected from the multi-trait meta-analysis were confrmed by bivariate association analysis. The previously reported pleiotropic efects of variants within the DGAT1 and MGST1 genes on three milk production traits, and pleiotropic efects of variants in GHR on milk yield and fat yield were con- frmed. -

Nuclear Factor I/B Is an Oncogene in Small Cell Lung Cancer
Nuclear Factor I/B is an Oncogene in Small Cell Lung Cancer The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Dooley, A. L. et al. “Nuclear factor I/B is an oncogene in small cell lung cancer.” Genes & Development 25 (2011): 1470-1475. As Published http://dx.doi.org/10.1101/gad.2046711 Publisher Cold Spring Harbor Laboratory Press in association with The Genetics Society Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/66512 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike 3.0 Detailed Terms http://creativecommons.org/licenses/by-nc-sa/3.0/ Dooley 1 Nuclear Factor I/B is an Oncogene in Small Cell Lung Cancer Alison L. Dooley1, Monte M. Winslow1, Derek Y. Chiang2,3,4, Shantanu Banerji2,3, Nicolas Stransky2, Talya L. Dayton1, Eric L. Snyder1, Stephanie Senna1, Charles A. Whittaker1, Roderick T. Bronson5, Denise Crowley1, Jordi Barretina2,3, Levi Garraway2,3, Matthew Meyerson2,3, Tyler Jacks1,6 1David H. Koch Institute for Integrative Cancer Research and Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA 2The Broad Institute, Cancer Program, Cambridge, Massachusetts, USA 3Dana-Farber Cancer Institute, Department of Medical Oncology and Center for Cancer Genome Discovery, Boston, Massachusetts, USA 4Current address: Lineberger Comprehensive Cancer Center, 450 West Drive, CB #7295, Chapel Hill, North Carolina, USA 5Department of Pathology, Tufts University School of Medicine and Veterinary Medicine, North Grafton, Massachusetts, USA 6Howard Hughes Medical Institute, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA Key Words: Small Cell Lung Cancer, Mouse model, Nuclear Factor I/B Dooley 2 Abstract Small cell lung cancer (SCLC) is an aggressive cancer often diagnosed after it has metastasized. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Original Article a Database and Functional Annotation of NF-Κb Target Genes
Int J Clin Exp Med 2016;9(5):7986-7995 www.ijcem.com /ISSN:1940-5901/IJCEM0019172 Original Article A database and functional annotation of NF-κB target genes Yang Yang, Jian Wu, Jinke Wang The State Key Laboratory of Bioelectronics, Southeast University, Nanjing 210096, People’s Republic of China Received November 4, 2015; Accepted February 10, 2016; Epub May 15, 2016; Published May 30, 2016 Abstract: Backgrounds: The previous studies show that the transcription factor NF-κB always be induced by many inducers, and can regulate the expressions of many genes. The aim of the present study is to explore the database and functional annotation of NF-κB target genes. Methods: In this study, we manually collected the most complete listing of all NF-κB target genes identified to date, including the NF-κB microRNA target genes and built the database of NF-κB target genes with the detailed information of each target gene and annotated it by DAVID tools. Results: The NF-κB target genes database was established (http://tfdb.seu.edu.cn/nfkb/). The collected data confirmed that NF-κB maintains multitudinous biological functions and possesses the considerable complexity and diversity in regulation the expression of corresponding target genes set. The data showed that the NF-κB was a central regula- tor of the stress response, immune response and cellular metabolic processes. NF-κB involved in bone disease, immunological disease and cardiovascular disease, various cancers and nervous disease. NF-κB can modulate the expression activity of other transcriptional factors. Inhibition of IKK and IκBα phosphorylation, the decrease of nuclear translocation of p65 and the reduction of intracellular glutathione level determined the up-regulation or down-regulation of expression of NF-κB target genes. -

Genomic Study of RNA Polymerase II and III Snapc-Bound Promoters Reveals a Gene Transcribed by Both Enzymes and a Broad Use of Common Activators
Genomic Study of RNA Polymerase II and III SNAPc-Bound Promoters Reveals a Gene Transcribed by Both Enzymes and a Broad Use of Common Activators Nicole James Faresse1., Donatella Canella1., Viviane Praz1,2, Joe¨lle Michaud1¤, David Romascano1, Nouria Hernandez1* 1 Center for Integrative Genomics, Faculty of Biology and Medicine, University of Lausanne, Lausanne, Switzerland, 2 Swiss Institute of Bioinformatics, Lausanne, Switzerland Abstract SNAPc is one of a few basal transcription factors used by both RNA polymerase (pol) II and pol III. To define the set of active SNAPc-dependent promoters in human cells, we have localized genome-wide four SNAPc subunits, GTF2B (TFIIB), BRF2, pol II, and pol III. Among some seventy loci occupied by SNAPc and other factors, including pol II snRNA genes, pol III genes with type 3 promoters, and a few un-annotated loci, most are primarily occupied by either pol II and GTF2B, or pol III and BRF2. A notable exception is the RPPH1 gene, which is occupied by significant amounts of both polymerases. We show that the large majority of SNAPc-dependent promoters recruit POU2F1 and/or ZNF143 on their enhancer region, and a subset also recruits GABP, a factor newly implicated in SNAPc-dependent transcription. These activators associate with pol II and III promoters in G1 slightly before the polymerase, and ZNF143 is required for efficient transcription initiation complex assembly. The results characterize a set of genes with unique properties and establish that polymerase specificity is not absolute in vivo. Citation: James Faresse N, Canella D, Praz V, Michaud J, Romascano D, et al. -

Mapping of a Chromosome 12 Region Associated with Airway Hyperresponsiveness in a Recombinant Congenic Mouse Strain and Selectio
Mapping of a Chromosome 12 Region Associated with Airway Hyperresponsiveness in a Recombinant Congenic Mouse Strain and Selection of Potential Candidate Genes by Expression and Sequence Variation Analyses Cynthia Kanagaratham1*, Rafael Marino2, Pierre Camateros2, John Ren3, Daniel Houle4, Robert Sladek1,2,5, Silvia M. Vidal1,3, Danuta Radzioch1,2 1 Department of Human Genetics, McGill University, Montreal, Quebec, Canada, 2 Faculty of Medicine, Division of Experimental Medicine, McGill University, Montreal, Quebec, Canada, 3 Department of Microbiology and Immunology, McGill University, Montreal, Quebec, Canada, 4 Research Institute of the McGill University Health Center, Montreal, Quebec, Canada, 5 McGill University and Genome Quebec Innovation Centre, Montreal, Quebec, Canada Abstract In a previous study we determined that BcA86 mice, a strain belonging to a panel of AcB/BcA recombinant congenic strains, have an airway responsiveness phenotype resembling mice from the airway hyperresponsive A/J strain. The majority of the BcA86 genome is however from the hyporesponsive C57BL/6J strain. The aim of this study was to identify candidate regions and genes associated with airway hyperresponsiveness (AHR) by quantitative trait locus (QTL) analysis using the BcA86 strain. Airway responsiveness of 205 F2 mice generated from backcrossing BcA86 strain to C57BL/6J strain was measured and used for QTL analysis to identify genomic regions in linkage with AHR. Consomic mice for the QTL containing chromosomes were phenotyped to study the contribution of each chromosome to lung responsiveness. Candidate genes within the QTL were selected based on expression differences in mRNA from whole lungs, and the presence of coding non- synonymous mutations that were predicted to have a functional effect by amino acid substitution prediction tools. -

Genomic Anatomy of the Tyrp1 (Brown) Deletion Complex
Genomic anatomy of the Tyrp1 (brown) deletion complex Ian M. Smyth*, Laurens Wilming†, Angela W. Lee*, Martin S. Taylor*, Phillipe Gautier*, Karen Barlow†, Justine Wallis†, Sancha Martin†, Rebecca Glithero†, Ben Phillimore†, Sarah Pelan†, Rob Andrew†, Karen Holt†, Ruth Taylor†, Stuart McLaren†, John Burton†, Jonathon Bailey†, Sarah Sims†, Jan Squares†, Bob Plumb†, Ann Joy†, Richard Gibson†, James Gilbert†, Elizabeth Hart†, Gavin Laird†, Jane Loveland†, Jonathan Mudge†, Charlie Steward†, David Swarbreck†, Jennifer Harrow†, Philip North‡, Nicholas Leaves‡, John Greystrong‡, Maria Coppola‡, Shilpa Manjunath‡, Mark Campbell‡, Mark Smith‡, Gregory Strachan‡, Calli Tofts‡, Esther Boal‡, Victoria Cobley‡, Giselle Hunter‡, Christopher Kimberley‡, Daniel Thomas‡, Lee Cave-Berry‡, Paul Weston‡, Marc R. M. Botcherby‡, Sharon White*, Ruth Edgar*, Sally H. Cross*, Marjan Irvani¶, Holger Hummerich¶, Eleanor H. Simpson*, Dabney Johnson§, Patricia R. Hunsicker§, Peter F. R. Little¶, Tim Hubbard†, R. Duncan Campbell‡, Jane Rogers†, and Ian J. Jackson*ʈ *Medical Research Council Human Genetics Unit, Edinburgh EH4 2XU, United Kingdom; †Wellcome Trust Sanger Institute, and ‡Medical Research Council Rosalind Franklin Centre for Genome Research, Hinxton CB10 1SA, United Kingdom; §Life Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831; and ¶Department of Biochemistry, Imperial College, London SW7 2AZ, United Kingdom Communicated by Liane B. Russell, Oak Ridge National Laboratory, Oak Ridge, TN, January 9, 2006 (received for review September 15, 2005) Chromosome deletions in the mouse have proven invaluable in the deletions also provided the means to produce physical maps of dissection of gene function. The brown deletion complex com- genetic markers. Studies of this kind have been published for prises >28 independent genome rearrangements, which have several loci, including albino (Tyr), piebald (Ednrb), pink-eyed been used to identify several functional loci on chromosome 4 dilution (p), and the brown deletion complex (2–6). -

Genomic and Expression Profiling of Human Spermatocytic Seminomas: Primary Spermatocyte As Tumorigenic Precursor and DMRT1 As Candidate Chromosome 9 Gene
Research Article Genomic and Expression Profiling of Human Spermatocytic Seminomas: Primary Spermatocyte as Tumorigenic Precursor and DMRT1 as Candidate Chromosome 9 Gene Leendert H.J. Looijenga,1 Remko Hersmus,1 Ad J.M. Gillis,1 Rolph Pfundt,4 Hans J. Stoop,1 Ruud J.H.L.M. van Gurp,1 Joris Veltman,1 H. Berna Beverloo,2 Ellen van Drunen,2 Ad Geurts van Kessel,4 Renee Reijo Pera,5 Dominik T. Schneider,6 Brenda Summersgill,7 Janet Shipley,7 Alan McIntyre,7 Peter van der Spek,3 Eric Schoenmakers,4 and J. Wolter Oosterhuis1 1Department of Pathology, Josephine Nefkens Institute; Departments of 2Clinical Genetics and 3Bioinformatics, Erasmus Medical Center/ University Medical Center, Rotterdam, the Netherlands; 4Department of Human Genetics, Radboud University Medical Center, Nijmegen, the Netherlands; 5Howard Hughes Medical Institute, Whitehead Institute and Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts; 6Clinic of Paediatric Oncology, Haematology and Immunology, Heinrich-Heine University, Du¨sseldorf, Germany; 7Molecular Cytogenetics, Section of Molecular Carcinogenesis, The Institute of Cancer Research, Sutton, Surrey, United Kingdom Abstract histochemistry, DMRT1 (a male-specific transcriptional regulator) was identified as a likely candidate gene for Spermatocytic seminomas are solid tumors found solely in the involvement in the development of spermatocytic seminomas. testis of predominantly elderly individuals. We investigated these tumors using a genome-wide analysis for structural and (Cancer Res 2006; 66(1): 290-302) numerical chromosomal changes through conventional kar- yotyping, spectral karyotyping, and array comparative Introduction genomic hybridization using a 32 K genomic tiling-path Spermatocytic seminomas are benign testicular tumors that resolution BAC platform (confirmed by in situ hybridization).