Chromosome 3P Loss of Heterozygosity Is Associated with a Unique Metabolic Network in Clear Cell Renal Carcinoma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

KO Kidney.Xlsx
Supplemental Table 18: Dietary Impact on the CGL KO Kidney Sulfhydrome DR/AL Accession Molecular Cysteine Spectral Protein Name Number Alternate ID Weight Residues Count Ratio P‐value Ig gamma‐2A chain C region, A allele P01863 (+1) Ighg 36 kDa 10 C 5.952 0.03767 Heterogeneous nuclear ribonucleoprotein M Q9D0E1 (+1) Hnrnpm 78 kDa 6 C 5.000 0.00595 Phospholipase D3 O35405 Pld3 54 kDa 8 C 4.167 0.04761 Ig kappa chain V‐V region L7 (Fragment) P01642 Gm10881 13 kDa 2 C 2.857 0.01232 UPF0160 protein MYG1, mitochondrial Q9JK81 Myg1 43 kDa 7 C 2.333 0.01613 Copper homeostasis protein cutC homolog Q9D8X1 Cutc 29 kDa 7 C 10.333 0.16419 Corticosteroid‐binding globulin Q06770 Serpina6 45 kDa 3 C 10.333 0.16419 28S ribosomal protein S22, mitochondrial Q9CXW2 Mrps22 41 kDa 2 C 7.333 0.3739 Isoform 3 of Agrin A2ASQ1‐3 Agrn 198 kDa 2 C 7.333 0.3739 3‐oxoacyl‐[acyl‐carrier‐protein] synthase, mitochondrial Q9D404 Oxsm 49 kDa 11 C 7.333 0.3739 Cordon‐bleu protein‐like 1 Q3UMF0 (+3)Cobll1 137 kDa 10 C 5.833 0.10658 ADP‐sugar pyrophosphatase Q9JKX6 Nudt5 24 kDa 5 C 4.167 0.15819 Complement C4‐B P01029 C4b 193 kDa 29 C 3.381 0.23959 Protein‐glutamine gamma‐glutamyltransferase 2 P21981 Tgm2 77 kDa 20 C 3.381 0.23959 Isochorismatase domain‐containing protein 1 Q91V64 Isoc1 32 kDa 5 C 3.333 0.10588 Serpin B8 O08800 Serpinb8 42 kDa 11 C 2.903 0.06902 Heterogeneous nuclear ribonucleoprotein A0 Q9CX86 Hnrnpa0 31 kDa 3 C 2.667 0.5461 Proteasome subunit beta type‐8 P28063 Psmb8 30 kDa 5 C 2.583 0.36848 Ig kappa chain V‐V region MOPC 149 P01636 12 kDa 2 C 2.583 0.36848 -

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

IMPDH2: a New Gene Associated with Dominant Juvenile-Onset Dystonia-Tremor Disorder
www.nature.com/ejhg BRIEF COMMUNICATION OPEN IMPDH2: a new gene associated with dominant juvenile-onset dystonia-tremor disorder 1,8 1,8 2 3 1,4 2 5 Anna Kuukasjärvi , Juan✉ C. Landoni , Jyrki Kaukonen , Mika Juhakoski , Mari Auranen , Tommi Torkkeli , Vidya Velagapudi and Anu Suomalainen 1,6,7 © The Author(s) 2021 The aetiology of dystonia disorders is complex, and next-generation sequencing has become a useful tool in elucidating the variable genetic background of these diseases. Here we report a deleterious heterozygous truncating variant in the inosine monophosphate dehydrogenasegene(IMPDH2) by whole-exome sequencing, co-segregating with a dominantly inherited dystonia-tremor disease in a large Finnish family. We show that the defect results in degradation of the gene product, causing IMPDH2 deficiency in patient cells. IMPDH2 is the first and rate-limiting enzyme in the de novo biosynthesis of guanine nucleotides, a dopamine synthetic pathway previously linked to childhood or adolescence-onset dystonia disorders. We report IMPDH2 as a new gene to the dystonia disease entity. The evidence underlines the important link between guanine metabolism, dopamine biosynthesis and dystonia. European Journal of Human Genetics; https://doi.org/10.1038/s41431-021-00939-1 INTRODUCTION The disease-onset was between 9 and 20 years of age. Table 1 Dystonias are rare movement disorders characterised by sustained or summarises the clinical presentations. intermittent muscle contractions causing abnormal, often repetitive, movements and/or postures. Dystonia can manifest as an isolated Case report symptom or combined with e.g. parkinsonism or myoclonus [1]. While Patient II-6 is a 46-year-old woman. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Chuanxiong Rhizoma Compound on HIF-VEGF Pathway and Cerebral Ischemia-Reperfusion Injury’S Biological Network Based on Systematic Pharmacology
ORIGINAL RESEARCH published: 25 June 2021 doi: 10.3389/fphar.2021.601846 Exploring the Regulatory Mechanism of Hedysarum Multijugum Maxim.-Chuanxiong Rhizoma Compound on HIF-VEGF Pathway and Cerebral Ischemia-Reperfusion Injury’s Biological Network Based on Systematic Pharmacology Kailin Yang 1†, Liuting Zeng 1†, Anqi Ge 2†, Yi Chen 1†, Shanshan Wang 1†, Xiaofei Zhu 1,3† and Jinwen Ge 1,4* Edited by: 1 Takashi Sato, Key Laboratory of Hunan Province for Integrated Traditional Chinese and Western Medicine on Prevention and Treatment of 2 Tokyo University of Pharmacy and Life Cardio-Cerebral Diseases, Hunan University of Chinese Medicine, Changsha, China, Galactophore Department, The First 3 Sciences, Japan Hospital of Hunan University of Chinese Medicine, Changsha, China, School of Graduate, Central South University, Changsha, China, 4Shaoyang University, Shaoyang, China Reviewed by: Hui Zhao, Capital Medical University, China Background: Clinical research found that Hedysarum Multijugum Maxim.-Chuanxiong Maria Luisa Del Moral, fi University of Jaén, Spain Rhizoma Compound (HCC) has de nite curative effect on cerebral ischemic diseases, *Correspondence: such as ischemic stroke and cerebral ischemia-reperfusion injury (CIR). However, its Jinwen Ge mechanism for treating cerebral ischemia is still not fully explained. [email protected] †These authors share first authorship Methods: The traditional Chinese medicine related database were utilized to obtain the components of HCC. The Pharmmapper were used to predict HCC’s potential targets. Specialty section: The CIR genes were obtained from Genecards and OMIM and the protein-protein This article was submitted to interaction (PPI) data of HCC’s targets and IS genes were obtained from String Ethnopharmacology, a section of the journal database. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
Type of the Paper (Article
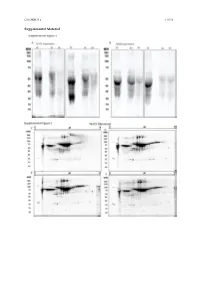
Cells 2020, 9, x 1 of 19 Supplemental Material Cells 2020, 9, x 2 of 19 Figure S1. Secretome enrichment: protocol optimization. 1D SDS-PAGE documentation of washing steps: Culture medium was substituted with FCS-free medium, which was changed every 2 h. The supernatants were then collected, and the proteins isolated and separated in 1D SDS-PAGE ((A) TK173 and (B) TK188). Proteins were stained with Flamingo fluorescent gel stain. Two-dimensional pattern of the proteins isolated from supernatant of TK173, (C) 2 h, (D) 4 h, (E) 6 h, and (F) 8 h after changing to FCS-free medium. (G) Cell secretome collected 24 h after elimination of the contaminating FCS-proteins with different washing steps. Proteins were stained with Flamingo fluorescent gel stain. Cells 2020, 9, x 3 of 19 Figure S2. 2-DE reference maps of secretomes; 150 μg proteins were loaded on an 11 cm IPG strip with a linear pH gradient PI 5–8 for IEF; 12% SDS-polyacrylamide gels were used for the second dimension. Proteins were stained with Flamingo fluorescent gel stain. Identified spots were assigned a number corresponding to that in their table. 2-DE maps from secretome of (A) TK173 control and (B) TGFβ1- treated ones. The 2-DE patterns revealed an alteration of secretome in stimulated TK173. Secretome patterns from TK173 treated with (C) ANG II and (D) PDGF. Cells 2020, 9, x 4 of 19 A Figure S3. Classification of the differentially expressed proteins upon ANG II, TGFβ1, or PDGF treatment in TK173. (A) Bar charts of the cellular component analyzed by STRAP biological function analysis in which the identified proteins from all treatments in both cell types are involved. -

Supplementary Methods
Supplementary methods Human lung tissues and tissue microarray (TMA) All human tissues were obtained from the Lung Cancer Specialized Program of Research Excellence (SPORE) Tissue Bank at the M.D. Anderson Cancer Center (Houston, TX). A collection of 26 lung adenocarcinomas and 24 non-tumoral paired tissues were snap-frozen and preserved in liquid nitrogen for total RNA extraction. For each tissue sample, the percentage of malignant tissue was calculated and the cellular composition of specimens was determined by histological examination (I.I.W.) following Hematoxylin-Eosin (H&E) staining. All malignant samples retained contained more than 50% tumor cells. Specimens resected from NSCLC stages I-IV patients who had no prior chemotherapy or radiotherapy were used for TMA analysis by immunohistochemistry. Patients who had smoked at least 100 cigarettes in their lifetime were defined as smokers. Samples were fixed in formalin, embedded in paraffin, stained with H&E, and reviewed by an experienced pathologist (I.I.W.). The 413 tissue specimens collected from 283 patients included 62 normal bronchial epithelia, 61 bronchial hyperplasias (Hyp), 15 squamous metaplasias (SqM), 9 squamous dysplasias (Dys), 26 carcinomas in situ (CIS), as well as 98 squamous cell carcinomas (SCC) and 141 adenocarcinomas. Normal bronchial epithelia, hyperplasia, squamous metaplasia, dysplasia, CIS, and SCC were considered to represent different steps in the development of SCCs. All tumors and lesions were classified according to the World Health Organization (WHO) 2004 criteria. The TMAs were prepared with a manual tissue arrayer (Advanced Tissue Arrayer ATA100, Chemicon International, Temecula, CA) using 1-mm-diameter cores in triplicate for tumors and 1.5 to 2-mm cores for normal epithelial and premalignant lesions. -

1 T CELL ACTIVATION TRIGGERS REVERSIBLE INOSINE-5'-MONOPHOSPHATE DEHYDROGENASE ASSEMBLY Krisna C. Duong-Ly 1, Yin-Ming Kuo 2
bioRxiv preprint doi: https://doi.org/10.1101/315929; this version posted May 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. T CELL ACTIVATION TRIGGERS REVERSIBLE INOSINE-5’-MONOPHOSPHATE DEHYDROGENASE ASSEMBLY Krisna C. Duong-Ly1, Yin-Ming Kuo2, Matthew C. Johnson3, Justin M. Kollman3, Jonathan Soboloff4, Glenn F. Rall5, Andrew J. Andrews2, and Jeffrey R. Peterson1 1Cancer Biology Program, Fox Chase Cancer Center, Philadelphia, PA 2Cancer Epigenetics Program, Fox Chase Cancer Center, Philadelphia, PA 3Department of Biochemistry, University of Washington, Seattle, WA 4Fels Institute for Cancer Research and Molecular Biology, Temple University School of Medicine, Philadelphia, PA 5Blood Cell Development and Function Program, Fox Chase Cancer Center, Philadelphia, PA Correspondence to Jeffrey R. Peterson: [email protected], Fox Chase Cancer Center, 333 Cottman Avenue, Philadelphia, PA 19111, ORCID: 0000-0002-0604-718X; Yin-Ming Kuo’s present address is Department of Radiology, University of Pennsylvania School of Medicine, Philadelphia, PA Short running title: IMPDH FILAMENT ASSEMBLY IN T CELLS Abbreviations used: IMP: inosine-5’-monophosphate IMPDH: inosine-5’-monophosphate dehydrogenase 1 bioRxiv preprint doi: https://doi.org/10.1101/315929; this version posted May 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Placental Transcriptome Adaptations to Maternal Nutrient Restriction in Sheep
International Journal of Molecular Sciences Article Placental Transcriptome Adaptations to Maternal Nutrient Restriction in Sheep Chelsie B. Steinhauser 1,†, Colleen A. Lambo 2,†, Katharine Askelson 1, Gregory W. Burns 3, Susanta K. Behura 4,5, Thomas E. Spencer 4, Fuller W. Bazer 1 and Michael Carey Satterfield 1,* 1 Department of Animal Science, Texas A & M University, College Station, TX 77843, USA; [email protected] (C.B.S.); [email protected] (K.A.); [email protected] (F.W.B.) 2 Department of Veterinary Physiology and Pharmacology, Texas A & M University, College Station, TX 77843, USA; [email protected] 3 Department of Obstetrics, Gynecology and Reproductive Biology, Michigan State University, Grand Rapids, MI 49503, USA; [email protected] 4 Division of Animal Sciences, University of Missouri, Columbia, MO 65211, USA; [email protected] (S.K.B.); [email protected] (T.E.S.) 5 Institute for Data Science and Informatics, University of Missouri, Columbia, MO 65211, USA * Correspondence: csatterfi[email protected]; Tel.: +1-979-845-6448 † These authors contributed equally to this work. Abstract: Placental development is modified in response to maternal nutrient restriction (NR), resulting in a spectrum of fetal growth rates. Pregnant sheep carrying singleton fetuses and fed either 100% (n = 8) or 50% (NR; n = 28) of their National Research Council (NRC) recommended intake from days 35–135 of pregnancy were used to elucidate placentome transcriptome alterations at both Citation: Steinhauser, C.B.; Lambo, day 70 and day 135. NR fetuses were further designated into upper (NR NonSGA; n = 7) and lower C.A.; Askelson, K.; Burns, G.W.; quartiles (NR SGA; n = 7) based on day 135 fetal weight. -

(12) Patent Application Publication (10) Pub. No.: US 2005/0260639 A1 Nakamura Et Al
US 2005O260639A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2005/0260639 A1 Nakamura et al. (43) Pub. Date: Nov. 24, 2005 (54) METHOD FOR DIAGNOSING PANCREATIC Related U.S. Application Data CANCER (63) Continuation-in-part of application No. PCT/JPO3/ 11817, filed on Sep. 17, 2003. (75) Inventors: Yusuke Nakamura, Yokohama-shi (JP); Toyomasa Katagiri, Shinagawa-ku (60) Provisional application No. 60/555,809, filed on Mar. 24, 2004. Provisional application No. 60/450,889, (JP); Hidewaki Nakagawa, filed on Feb. 28, 2003. Provisional application No. Shinagawa-ku (JP) 60/414,872, filed on Sep. 30, 2002. Publication Classification Correspondence Address: TOWNSEND AND TOWNSEND AND CREW, (51) Int. Cl. .................................................. C12O 1/68 LLP (52) U.S. Cl. .................................................................. 435/6 TWO EMBARCADERO CENTER EIGHTH FLOOR (57) ABSTRACT SAN FRANCISCO, CA 94111-3834 (US) Objective methods for detecting and diagnosing pancreatic cancer (PNC) are described herein. In one embodiment, the (73) Assignees: Oncotherapy Science, Inc., Kawasaki diagnostic method involves determining the expression level shi (JP); The University of Tokyo, Bun of PNC-associated gene that discriminates between PNC kyo-ku (JP) cells and normal cells. The present invention further pro vides methods of Screening for therapeutic agents useful in (21) Appl. No.: 11/090,739 the treatment of pancreatic cancer, methods of treating pancreatic cancer and method of vaccinating a Subject (22) Filed: Mar. 24, 2005 against pancreatic cancer. Patent Application Publication Nov. 24, 2005 Sheet 1 of 16 US 2005/0260639 A1 FG 1 Patent Application Publication Nov. 24, 2005 Sheet 2 of 16 US 2005/0260639 A1 s - to co on S 2 2 O C C C D C C C D C) () () O U O n.