Primepcr™Assay Validation Report
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

SNP Genotypes of Olfactory Receptor Genes Associated with Olfactory Ability in German Shepherd Dogs
SHORT COMMUNICATION doi: 10.1111/age.12389 SNP genotypes of olfactory receptor genes associated with olfactory ability in German Shepherd dogs † ‡ – M. Yang*, G.-J. Geng , W. Zhang , L. Cui§, H.-X. Zhang and J.-L. Zheng‡ † *Police-dog Technology Department, National Police University of China, Shenyang, Liaoning 110034, China. Technology Department, ‡ Shenyang Traffic Police Detachment, Shenyang, Liaoning 110001, China. Forensic Medicine Department, National Police University of China, Shenyang, Liaoning 110854, China. §Document Inspection Department, National Police University of China, Shenyang, Liaoning – 110854, China. Mark Inspection Department, National Police University of China, Shenyang, Liaoning 110854, China. Summary To find out the relationship between SNP genotypes of canine olfactory receptor genes and olfactory ability, 28 males and 20 females from German Shepherd dogs in police service were scored by odor detection tests and analyzed using the Beckman GenomeLab SNPstream. The representative 22 SNP loci from the exonic regions of 12 olfactory receptor genes were investigated, and three kinds of odor (human, ice drug and trinitrotoluene) were detected. The results showed that the SNP genotypes at the OR10H1-like:c.632C>T, OR10H1-like:c.770A>T, OR2K2-like:c.518G>A, OR4C11-like: c.511T>G and OR4C11-like:c.692G>A loci had a statistically significant effect on the scenting abilities (P < 0.001). The kind of odor influenced the performances of the dogs (P < 0.001). In addition, there were interactions between genotype and the kind of odor at the following loci: OR10H1-like:c.632C>T, OR10H1-like:c.770A>T, OR4C11-like:c.511T>G and OR4C11-like:c.692G>A(P< 0.001). -

Genome-Wide Profiling of Druggable Active Tumor Defense Mechanisms to Enhance Cancer Immunotherapy
bioRxiv preprint doi: https://doi.org/10.1101/843185; this version posted November 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Genome-wide profiling of druggable active tumor defense mechanisms to enhance cancer immunotherapy Rigel J. Kishton1,2,*,#, Shashank J. Patel1,2,†,*, Suman K. Vodnala1,2, Amy E. Decker3, Yogin Patel1,2, Madhusudhanan Sukumar1,2, Tori N. Yamamoto1,2,4, Zhiya Yu1,2, Michelle Ji1,2, Amanda N. Henning1,2, Devikala Gurusamy1,2, Douglas C. Palmer1,2, Winifred Lo1, Anna Pasetto1, Parisa Malekzadeh1, Drew C. Deniger1, Kris C. Wood3, Neville E. Sanjana5,6, Nicholas P. Restifo1,2, #, § 1Surgery Branch, Center for Cancer Research, National Cancer Institute, Bethesda, MD 20892, USA 2Center for Cell-Based Therapy, National Cancer Institute, Bethesda, MD 20892, USA 3Department of Pharmacology & Cancer Biology, Duke University School of Medicine, Durham, NC, USA 4Immunology Graduate Group, University of Pennsylvania, Philadelphia, PA 19104, USA 5New York Genome Center, New York, NY 10013 USA 6Department of Biology, New York University, New York, NY 10003, USA *These authors contributed equally to this work. †Present address: NextCure Inc., Beltsville, MD 20705, USA §Present address: Lyell Immunopharma, South San Francisco, CA 94080, USA #Corresponding authors. NPR: [email protected]. RJK: [email protected]. bioRxiv preprint doi: https://doi.org/10.1101/843185; this version posted November 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. -

Clinical, Molecular, and Immune Analysis of Dabrafenib-Trametinib
Supplementary Online Content Chen G, McQuade JL, Panka DJ, et al. Clinical, molecular and immune analysis of dabrafenib-trametinib combination treatment for metastatic melanoma that progressed during BRAF inhibitor monotherapy: a phase 2 clinical trial. JAMA Oncology. Published online April 28, 2016. doi:10.1001/jamaoncol.2016.0509. eMethods. eReferences. eTable 1. Clinical efficacy eTable 2. Adverse events eTable 3. Correlation of baseline patient characteristics with treatment outcomes eTable 4. Patient responses and baseline IHC results eFigure 1. Kaplan-Meier analysis of overall survival eFigure 2. Correlation between IHC and RNAseq results eFigure 3. pPRAS40 expression and PFS eFigure 4. Baseline and treatment-induced changes in immune infiltrates eFigure 5. PD-L1 expression eTable 5. Nonsynonymous mutations detected by WES in baseline tumors This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/30/2021 eMethods Whole exome sequencing Whole exome capture libraries for both tumor and normal samples were constructed using 100ng genomic DNA input and following the protocol as described by Fisher et al.,3 with the following adapter modification: Illumina paired end adapters were replaced with palindromic forked adapters with unique 8 base index sequences embedded within the adapter. In-solution hybrid selection was performed using the Illumina Rapid Capture Exome enrichment kit with 38Mb target territory (29Mb baited). The targeted region includes 98.3% of the intervals in the Refseq exome database. Dual-indexed libraries were pooled into groups of up to 96 samples prior to hybridization. -

Positive Selection, Relaxation, and Acceleration in the Evolution of the Human and Chimp Genome
Positive Selection, Relaxation, and Acceleration in the Evolution of the Human and Chimp Genome Leonardo Arbiza1, Joaquı´n Dopazo2, Herna´n Dopazo1* 1 Pharmacogenomics and Comparative Genomics Unit, Centro de Investigacio´nPrı´ncipe Felipe (CIPF), Valencia, Spain, 2 Functional Genomics Unit, Bioinformatics Department, Centro de Investigacio´nPrı´ncipe Felipe (CIPF), Valencia, Spain For years evolutionary biologists have been interested in searching for the genetic bases underlying humanness. Recent efforts at a large or a complete genomic scale have been conducted to search for positively selected genes in human and in chimp. However, recently developed methods allowing for a more sensitive and controlled approach in the detection of positive selection can be employed. Here, using 13,198 genes, we have deduced the sets of genes involved in rate acceleration, positive selection, and relaxation of selective constraints in human, in chimp, and in their ancestral lineage since the divergence from murids. Significant deviations from the strict molecular clock were observed in 469 human and in 651 chimp genes. The more stringent branch-site test of positive selection detected 108 human and 577 chimp positively selected genes. An important proportion of the positively selected genes did not show a significant acceleration in rates, and similarly, many of the accelerated genes did not show significant signals of positive selection. Functional differentiation of genes under rate acceleration, positive selection, and relaxation was not statistically significant between human and chimp with the exception of terms related to G-protein coupled receptors and sensory perception. Both of these were over-represented under relaxation in human in relation to chimp. -

The Mouse Solitary Odorant Receptor Gene Promoters As Models for the Study of Odorant Receptor Gene Choice
RESEARCH ARTICLE The Mouse Solitary Odorant Receptor Gene Promoters as Models for the Study of Odorant Receptor Gene Choice Andrea Degl'Innocenti1,2*, Marta Parrilla1☯, Bettina Harr3☯, Meike Teschke3☯ 1 Max-Planck-Institut für Biophysik, Frankfurt am Main, Germany, 2 Unità di Biologia Cellulare e dello Sviluppo, Dipartimento di Biologia, Università di Pisa, Pisa, Italy, 3 Abteilung Evolutionsgenetik, Max-Planck- Institut für Evolutionsbiologie, Plön, Germany ☯ These authors contributed equally to this work. * [email protected] Abstract OPEN ACCESS Citation: Degl'Innocenti A, Parrilla M, Harr B, Background Teschke M (2016) The Mouse Solitary Odorant In vertebrates, several anatomical regions located within the nasal cavity mediate olfaction. Receptor Gene Promoters as Models for the Study of Odorant Receptor Gene Choice. PLoS ONE 11(1): Among these, the main olfactory epithelium detects most conventional odorants. Olfactory e0144698. doi:10.1371/journal.pone.0144698 sensory neurons, provided with cilia exposed to the air, detect volatile chemicals via an Editor: Johannes Reisert, Monell Chemical Senses extremely large family of seven-transmembrane chemoreceptors named odorant receptors. Center, UNITED STATES Their genes are expressed in a monogenic and monoallelic fashion: a single allele of a sin- Received: June 26, 2015 gle odorant receptor gene is transcribed in a given mature neuron, through a still uncharac- terized molecular mechanism known as odorant receptor gene choice. Accepted: November 23, 2015 Published: January 21, 2016 Copyright: © 2016 Degl'Innocenti et al. This is an Aim open access article distributed under the terms of the Odorant receptor genes are typically arranged in genomic clusters, but a few are isolated Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any (we call them solitary) from the others within a region broader than 1 Mb upstream and medium, provided the original author and source are downstream with respect to their transcript's coordinates. -

The Hypothalamus As a Hub for SARS-Cov-2 Brain Infection and Pathogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis Sreekala Nampoothiri1,2#, Florent Sauve1,2#, Gaëtan Ternier1,2ƒ, Daniela Fernandois1,2 ƒ, Caio Coelho1,2, Monica ImBernon1,2, Eleonora Deligia1,2, Romain PerBet1, Vincent Florent1,2,3, Marc Baroncini1,2, Florence Pasquier1,4, François Trottein5, Claude-Alain Maurage1,2, Virginie Mattot1,2‡, Paolo GiacoBini1,2‡, S. Rasika1,2‡*, Vincent Prevot1,2‡* 1 Univ. Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, DistAlz, UMR-S 1172, Lille, France 2 LaBoratorY of Development and PlasticitY of the Neuroendocrine Brain, FHU 1000 daYs for health, EGID, School of Medicine, Lille, France 3 Nutrition, Arras General Hospital, Arras, France 4 Centre mémoire ressources et recherche, CHU Lille, LiCEND, Lille, France 5 Univ. Lille, CNRS, INSERM, CHU Lille, Institut Pasteur de Lille, U1019 - UMR 8204 - CIIL - Center for Infection and ImmunitY of Lille (CIIL), Lille, France. # and ƒ These authors contriButed equallY to this work. ‡ These authors directed this work *Correspondence to: [email protected] and [email protected] Short title: Covid-19: the hypothalamic hypothesis 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -
Explorations in Olfactory Receptor Structure and Function by Jianghai
Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 ABSTRACT Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 Copyright by Jianghai Ho 2014 Abstract Olfaction is one of the most primitive of our senses, and the olfactory receptors that mediate this very important chemical sense comprise the largest family of genes in the mammalian genome. It is therefore surprising that we understand so little of how olfactory receptors work. In particular we have a poor idea of what chemicals are detected by most of the olfactory receptors in the genome, and for those receptors which we have paired with ligands, we know relatively little about how the structure of these ligands can either activate or inhibit the activation of these receptors. Furthermore the large repertoire of olfactory receptors, which belong to the G protein coupled receptor (GPCR) superfamily, can serve as a model to contribute to our broader understanding of GPCR-ligand binding, especially since GPCRs are important pharmaceutical targets. -

Investigating the Effects of Copy Number Variants on Reading and Language Performance Alessandro Gialluisi1,2, Alessia Visconti3, Erik G
Gialluisi et al. Journal of Neurodevelopmental Disorders (2016) 8:17 DOI 10.1186/s11689-016-9147-8 RESEARCH Open Access Investigating the effects of copy number variants on reading and language performance Alessandro Gialluisi1,2, Alessia Visconti3, Erik G. Willcutt4,5, Shelley D. Smith6, Bruce F. Pennington7, Mario Falchi3, John C. DeFries4,5, Richard K. Olson4,5, Clyde Francks1,8* and Simon E. Fisher1,8* Abstract Background: Reading and language skills have overlapping genetic bases, most of which are still unknown. Part of the missing heritability may be caused by copy number variants (CNVs). Methods: In a dataset of children recruited for a history of reading disability (RD, also known as dyslexia) or attention deficit hyperactivity disorder (ADHD) and their siblings, we investigated the effects of CNVs on reading and language performance. First, we called CNVs with PennCNV using signal intensity data from Illumina OmniExpress arrays (~723,000 probes). Then, we computed the correlation between measures of CNV genomic burden and the first principal component (PC) score derived from several continuous reading and language traits, both before and after adjustment for performance IQ. Finally, we screened the genome, probe-by-probe, for association with the PC scores, through two complementary analyses: we tested a binary CNV state assigned for the location of each probe (i.e., CNV+ or CNV−), and we analyzed continuous probe intensity data using FamCNV. Results: No significant correlation was found between measures of CNV burden and PC scores, and no genome-wide significant associations were detected in probe-by-probe screening. Nominally significant associations were detected (p~10−2–10−3)withinCNTN4 (contactin 4) and CTNNA3 (catenin alpha 3). -
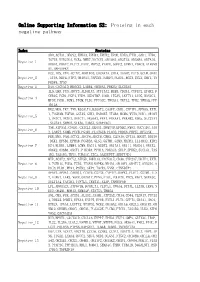
Online Supporting Information S2: Proteins in Each Negative Pathway
Online Supporting Information S2: Proteins in each negative pathway Index Proteins ADO,ACTA1,DEGS2,EPHA3,EPHB4,EPHX2,EPOR,EREG,FTH1,GAD1,HTR6, IGF1R,KIR2DL4,NCR3,NME7,NOTCH1,OR10S1,OR2T33,OR56B4,OR7A10, Negative_1 OR8G1,PDGFC,PLCZ1,PROC,PRPS2,PTAFR,SGPP2,STMN1,VDAC3,ATP6V0 A1,MAPKAPK2 DCC,IDS,VTN,ACTN2,AKR1B10,CACNA1A,CHIA,DAAM2,FUT5,GCLM,GNAZ Negative_2 ,ITPA,NEU4,NTF3,OR10A3,PAPSS1,PARD3,PLOD1,RGS3,SCLY,SHC1,TN FRSF4,TP53 Negative_3 DAO,CACNA1D,HMGCS2,LAMB4,OR56A3,PRKCQ,SLC25A5 IL5,LHB,PGD,ADCY3,ALDH1A3,ATP13A2,BUB3,CD244,CYFIP2,EPHX2,F CER1G,FGD1,FGF4,FZD9,HSD17B7,IL6R,ITGAV,LEFTY1,LIPG,MAN1C1, Negative_4 MPDZ,PGM1,PGM3,PIGM,PLD1,PPP3CC,TBXAS1,TKTL2,TPH2,YWHAQ,PPP 1R12A HK2,MOS,TKT,TNN,B3GALT4,B3GAT3,CASP7,CDH1,CYFIP1,EFNA5,EXTL 1,FCGR3B,FGF20,GSTA5,GUK1,HSD3B7,ITGB4,MCM6,MYH3,NOD1,OR10H Negative_5 1,OR1C1,OR1E1,OR4C11,OR56A3,PPA1,PRKAA1,PRKAB2,RDH5,SLC27A1 ,SLC2A4,SMPD2,STK36,THBS1,SERPINC1 TNR,ATP5A1,CNGB1,CX3CL1,DEGS1,DNMT3B,EFNB2,FMO2,GUCY1B3,JAG Negative_6 2,LARS2,NUMB,PCCB,PGAM1,PLA2G1B,PLOD2,PRDX6,PRPS1,RFXANK FER,MVD,PAH,ACTC1,ADCY4,ADCY8,CBR3,CLDN16,CPT1A,DDOST,DDX56 ,DKK1,EFNB1,EPHA8,FCGR3A,GLS2,GSTM1,GZMB,HADHA,IL13RA2,KIR2 Negative_7 DS4,KLRK1,LAMB4,LGMN,MAGI1,NUDT2,OR13A1,OR1I1,OR4D11,OR4X2, OR6K2,OR8B4,OXCT1,PIK3R4,PPM1A,PRKAG3,SELP,SPHK2,SUCLG1,TAS 1R2,TAS1R3,THY1,TUBA1C,ZIC2,AASDHPPT,SERPIND1 MTR,ACAT2,ADCY2,ATP5D,BMPR1A,CACNA1E,CD38,CYP2A7,DDIT4,EXTL Negative_8 1,FCER1G,FGD3,FZD5,ITGAM,MAPK8,NR4A1,OR10V1,OR4F17,OR52D1,O R8J3,PLD1,PPA1,PSEN2,SKP1,TACR3,VNN1,CTNNBIP1 APAF1,APOA1,CARD11,CCDC6,CSF3R,CYP4F2,DAPK1,FLOT1,GSTM1,IL2 -

Increased Genomic Burden of Germline Copy Number Variants Is Associated with Early Onset Breast Cancer: Australian Breast Cancer Family Registry Logan C
Walker et al. Breast Cancer Research (2017) 19:30 DOI 10.1186/s13058-017-0825-6 RESEARCH ARTICLE Open Access Increased genomic burden of germline copy number variants is associated with early onset breast cancer: Australian breast cancer family registry Logan C. Walker1, John F. Pearson2, George A. R. Wiggins1, Graham G. Giles3, John L. Hopper4* and Melissa C. Southey5 Abstract Background: Women with breast cancer who have multiple affected relatives are more likely to have inherited genetic risk factors for the disease. All the currently known genetic risk factors for breast cancer account for less than half of the average familial risk. Furthermore, the genetic factor(s) underlying an increased cancer risk for many women from multiple-case families remain unknown. Rare genomic duplications and deletions, known as copy number variants (CNVs), cover more than 10% of a human genome, are often not assessed in studies of genetic predisposition, and could account for some of the so-called “missing heritability”. Methods: We carried out a hypothesis-generating case-control study of breast cancer diagnosed before age 40 years (200 cases, 293 controls) using population-based cases from the Australian Breast Cancer Family Study. Genome-wide scanning for CNVs was performed using the Human610-Quad BeadChip and fine-mapping was conducted using PennCNV. Results: We identified deletions overlapping two known cancer susceptibility genes, (BRCA1 and BLM), and a duplication overlapping SMARCB1, associated with risk. The number of deletions across the genome was 1.5-fold higher for cases than controls (P =10-16), and 2-fold higher when only rare deletions overlapping genes (frequency <1%) were assessed (P =5×10-4). -

A Point of Rarity in Genetic Risk for Bipolar Disorder and Schizophrenia
WEB-ONLY CONTENT Rare Copy Number Variants A Point of Rarity in Genetic Risk for Bipolar Disorder and Schizophrenia Detelina Grozeva, MSc; George Kirov, PhD, MRCPsych; Dobril Ivanov, MSc; Ian R. Jones, PhD, MRCPsych; Lisa Jones, PhD; Elaine K. Green, PhD; David M. St Clair, MD, PhD; Allan H. Young, PhD, FRCPsych; Nicol Ferrier, PhD, FRCPsych; Anne E. Farmer, PhD, FRCPsych; Peter McGuffin, PhD, FRCPsych; Peter A. Holmans, PhD*; Michael J. Owen, PhD, FRCPsych*; Michael C. O’Donovan, PhD, FRCPsych*; Nick Craddock, PhD, FRCPsych*; for the Wellcome Trust Case Control Consortium Arch Gen Psychiatry. 2010;67(4):318-327 14 RESULTS schizophrenia article. In cases, this is caused by the ex- clusion of schizoaffective cases in the current analysis. The differences with regard to controls are caused by re- COMPARISON OF BURDEN OF COPY NUMBER analysis of a small subset of them. Some of the control VARIANTS ACCORDING TO SIZE BETWEEN data were processed with the use of different reference BIPOLAR CASES, CONTROLS, AND batches, which enabled us to include more controls in SCHIZOPHRENIA CASES the current analysis. We compared our bipolar disorder cases against our set of schizophrenia cases from the same population who had CNVs THAT OCCURRED MORE OFTEN IN been examined by the same methods (n=440).14 The copy BIPOLAR DISORDER CASES THAN CONTROLS number variants (CNVs) were classified into size cat- egories as in our previous report. eTable 1 shows the Although we did not observe an overall increase of CNV results. Compared with bipolar disorder, in the schizo- burden in bipolar cases compared with controls, some phrenia sample we observed a significant excess of large individual CNVs were more common in cases than con- deletions (PϽ.001) and total large CNVs (PϽ.001) and trols.