Genetic Analysis of Very Obese Children with Autism Spectrum Disorder
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

SNP Genotypes of Olfactory Receptor Genes Associated with Olfactory Ability in German Shepherd Dogs
SHORT COMMUNICATION doi: 10.1111/age.12389 SNP genotypes of olfactory receptor genes associated with olfactory ability in German Shepherd dogs † ‡ – M. Yang*, G.-J. Geng , W. Zhang , L. Cui§, H.-X. Zhang and J.-L. Zheng‡ † *Police-dog Technology Department, National Police University of China, Shenyang, Liaoning 110034, China. Technology Department, ‡ Shenyang Traffic Police Detachment, Shenyang, Liaoning 110001, China. Forensic Medicine Department, National Police University of China, Shenyang, Liaoning 110854, China. §Document Inspection Department, National Police University of China, Shenyang, Liaoning – 110854, China. Mark Inspection Department, National Police University of China, Shenyang, Liaoning 110854, China. Summary To find out the relationship between SNP genotypes of canine olfactory receptor genes and olfactory ability, 28 males and 20 females from German Shepherd dogs in police service were scored by odor detection tests and analyzed using the Beckman GenomeLab SNPstream. The representative 22 SNP loci from the exonic regions of 12 olfactory receptor genes were investigated, and three kinds of odor (human, ice drug and trinitrotoluene) were detected. The results showed that the SNP genotypes at the OR10H1-like:c.632C>T, OR10H1-like:c.770A>T, OR2K2-like:c.518G>A, OR4C11-like: c.511T>G and OR4C11-like:c.692G>A loci had a statistically significant effect on the scenting abilities (P < 0.001). The kind of odor influenced the performances of the dogs (P < 0.001). In addition, there were interactions between genotype and the kind of odor at the following loci: OR10H1-like:c.632C>T, OR10H1-like:c.770A>T, OR4C11-like:c.511T>G and OR4C11-like:c.692G>A(P< 0.001). -

List of Genes Associated with Sudden Cardiac Death (Scdgseta) Gene
List of genes associated with sudden cardiac death (SCDgseta) mRNA expression in normal human heart Entrez_I Gene symbol Gene name Uniprot ID Uniprot name fromb D GTEx BioGPS SAGE c d e ATP-binding cassette subfamily B ABCB1 P08183 MDR1_HUMAN 5243 √ √ member 1 ATP-binding cassette subfamily C ABCC9 O60706 ABCC9_HUMAN 10060 √ √ member 9 ACE Angiotensin I–converting enzyme P12821 ACE_HUMAN 1636 √ √ ACE2 Angiotensin I–converting enzyme 2 Q9BYF1 ACE2_HUMAN 59272 √ √ Acetylcholinesterase (Cartwright ACHE P22303 ACES_HUMAN 43 √ √ blood group) ACTC1 Actin, alpha, cardiac muscle 1 P68032 ACTC_HUMAN 70 √ √ ACTN2 Actinin alpha 2 P35609 ACTN2_HUMAN 88 √ √ √ ACTN4 Actinin alpha 4 O43707 ACTN4_HUMAN 81 √ √ √ ADRA2B Adrenoceptor alpha 2B P18089 ADA2B_HUMAN 151 √ √ AGT Angiotensinogen P01019 ANGT_HUMAN 183 √ √ √ AGTR1 Angiotensin II receptor type 1 P30556 AGTR1_HUMAN 185 √ √ AGTR2 Angiotensin II receptor type 2 P50052 AGTR2_HUMAN 186 √ √ AKAP9 A-kinase anchoring protein 9 Q99996 AKAP9_HUMAN 10142 √ √ √ ANK2/ANKB/ANKYRI Ankyrin 2 Q01484 ANK2_HUMAN 287 √ √ √ N B ANKRD1 Ankyrin repeat domain 1 Q15327 ANKR1_HUMAN 27063 √ √ √ ANKRD9 Ankyrin repeat domain 9 Q96BM1 ANKR9_HUMAN 122416 √ √ ARHGAP24 Rho GTPase–activating protein 24 Q8N264 RHG24_HUMAN 83478 √ √ ATPase Na+/K+–transporting ATP1B1 P05026 AT1B1_HUMAN 481 √ √ √ subunit beta 1 ATPase sarcoplasmic/endoplasmic ATP2A2 P16615 AT2A2_HUMAN 488 √ √ √ reticulum Ca2+ transporting 2 AZIN1 Antizyme inhibitor 1 O14977 AZIN1_HUMAN 51582 √ √ √ UDP-GlcNAc: betaGal B3GNT7 beta-1,3-N-acetylglucosaminyltransfe Q8NFL0 -

Heterotrimeric Go Protein Links Wnt-Frizzled Signaling with Ankyrins to Regulate the Neuronal Microtubule Cytoskeleton Anne-Marie Lüchtenborg1,2, Gonzalo P
© 2014. Published by The Company of Biologists Ltd | Development (2014) 141, 3399-3409 doi:10.1242/dev.106773 RESEARCH ARTICLE Heterotrimeric Go protein links Wnt-Frizzled signaling with ankyrins to regulate the neuronal microtubule cytoskeleton Anne-Marie Lüchtenborg1,2, Gonzalo P. Solis1, Diane Egger-Adam2, Alexey Koval1, Chen Lin1,2, Maxime G. Blanchard1, Stephan Kellenberger1 and Vladimir L. Katanaev1,2,* ABSTRACT The evolutionarily conserved Wg pathway is important for Drosophila neuromuscular junctions (NMJs) represent a powerful numerous developmental programs and cellular processes (Logan model system with which to study glutamatergic synapse formation and Nusse, 2004). In the nervous system of Drosophila,Wg and remodeling. Several proteins have been implicated in these signaling is involved in the formation of neuromuscular junctions processes, including components of canonical Wingless (Drosophila (NMJs) (Packard et al., 2002; Miech et al., 2008). Being a Wnt1) signaling and the giant isoforms of the membrane-cytoskeleton glutamatergic synapse, the Drosophila NMJ provides a useful linker Ankyrin 2, but possible interconnections and cooperation experimental model with which to study mammalian central between these proteins were unknown. Here, we demonstrate that nervous system synapses, their formation and remodeling (Collins the heterotrimeric G protein Go functions as a transducer of Wingless- and DiAntonio, 2007). The Drosophila NMJ is a beads-on-a-string- Frizzled 2 signaling in the synapse. We identify Ankyrin 2 as a target like structure that is formed at the axon terminus and is composed of – – of Go signaling required for NMJ formation. Moreover, the Go-ankyrin distinct circular structures the synaptic boutons which contain interaction is conserved in the mammalian neurite outgrowth pathway. -

Circular RNA Hsa Circ 0005114‑Mir‑142‑3P/Mir‑590‑5P‑ Adenomatous
ONCOLOGY LETTERS 21: 58, 2021 Circular RNA hsa_circ_0005114‑miR‑142‑3p/miR‑590‑5p‑ adenomatous polyposis coli protein axis as a potential target for treatment of glioma BO WEI1*, LE WANG2* and JINGWEI ZHAO1 1Department of Neurosurgery, China‑Japan Union Hospital of Jilin University, Changchun, Jilin 130033; 2Department of Ophthalmology, The First Hospital of Jilin University, Jilin University, Changchun, Jilin 130021, P.R. China Received September 12, 2019; Accepted October 22, 2020 DOI: 10.3892/ol.2020.12320 Abstract. Glioma is the most common type of brain tumor APC expression with a good overall survival rate. UALCAN and is associated with a high mortality rate. Despite recent analysis using TCGA data of glioblastoma multiforme and the advances in treatment options, the overall prognosis in patients GSE25632 and GSE103229 microarray datasets showed that with glioma remains poor. Studies have suggested that circular hsa‑miR‑142‑3p/hsa‑miR‑590‑5p was upregulated and APC (circ)RNAs serve important roles in the development and was downregulated. Thus, hsa‑miR‑142‑3p/hsa‑miR‑590‑5p‑ progression of glioma and may have potential as therapeutic APC‑related circ/ceRNA axes may be important in glioma, targets. However, the expression profiles of circRNAs and their and hsa_circ_0005114 interacted with both of these miRNAs. functions in glioma have rarely been studied. The present study Functional analysis showed that hsa_circ_0005114 was aimed to screen differentially expressed circRNAs (DECs) involved in insulin secretion, while APC was associated with between glioma and normal brain tissues using sequencing the Wnt signaling pathway. In conclusion, hsa_circ_0005114‑ data collected from the Gene Expression Omnibus database miR‑142‑3p/miR‑590‑5p‑APC ceRNA axes may be potential (GSE86202 and GSE92322 datasets) and explain their mecha‑ targets for the treatment of glioma. -

Cardiomyopathy
JACC: BASIC TO TRANSLATIONAL SCIENCE VOL.1,NO.5,2016 ª 2016 THE AUTHORS. PUBLISHED BY ELSEVIER ON BEHALF OF THE AMERICAN ISSN 2452-302X COLLEGE OF CARDIOLOGY FOUNDATION. THIS IS AN OPEN ACCESS ARTICLE UNDER http://dx.doi.org/10.1016/j.jacbts.2016.05.004 THE CC BY-NC-ND LICENSE (http://creativecommons.org/licenses/by-nc-nd/4.0/). PRE-CLINICAL RESEARCH FLNC Gene Splice Mutations Cause Dilated Cardiomyopathy a a b,c a d Rene L. Begay, BS, Charles A. Tharp, MD, August Martin, Sharon L. Graw, PHD, Gianfranco Sinagra, MD, e a a b,c,f b,c Daniela Miani, MD, Mary E. Sweet, BA, Dobromir B. Slavov, PHD, Neil Stafford, MD, Molly J. Zeller, b,c a d g g Rasha Alnefaie, Teisha J. Rowland, PHD, Francesca Brun, MD, Kenneth L. Jones, PHD, Katherine Gowan, a b,c a Luisa Mestroni, MD, Deborah M. Garrity, PHD, Matthew R.G. Taylor, MD, PHD VISUAL ABSTRACT HIGHLIGHTS Deoxyribonucleic acid obtained from 2 large DCM families was studied using whole-exome sequencing and cose- gregation analysis resulting in the iden- tification of a novel disease gene, FLNC. The2families,fromthesameItalian region, harbored the same FLNC splice- site mutation (FLNC c.7251D1G>A). A third U.S. family was then identified with a novel FLNC splice-site mutation (FLNC c.5669-1delG) that leads to haploinsufficiency as shown by the FLNC Western blot analysis of the heart muscle. The FLNC ortholog flncb morpholino was injected into zebrafish embryos, and when flncb was knocked down caused a cardiac dysfunction phenotype. -

Copy Number Variation in Fetal Alcohol Spectrum Disorder
Biochemistry and Cell Biology Copy number variation in fetal alcohol spectrum disorder Journal: Biochemistry and Cell Biology Manuscript ID bcb-2017-0241.R1 Manuscript Type: Article Date Submitted by the Author: 09-Nov-2017 Complete List of Authors: Zarrei, Mehdi; The Centre for Applied Genomics Hicks, Geoffrey G.; University of Manitoba College of Medicine, Regenerative Medicine Reynolds, James N.; Queen's University School of Medicine, Biomedical and Molecular SciencesDraft Thiruvahindrapuram, Bhooma; The Centre for Applied Genomics Engchuan, Worrawat; Hospital for Sick Children SickKids Learning Institute Pind, Molly; University of Manitoba College of Medicine, Regenerative Medicine Lamoureux, Sylvia; The Centre for Applied Genomics Wei, John; The Centre for Applied Genomics Wang, Zhouzhi; The Centre for Applied Genomics Marshall, Christian R.; The Centre for Applied Genomics Wintle, Richard; The Centre for Applied Genomics Chudley, Albert; University of Manitoba Scherer, Stephen W.; The Centre for Applied Genomics Is the invited manuscript for consideration in a Special Fetal Alcohol Spectrum Disorder Issue? : Keyword: Fetal alcohol spectrum disorder, FASD, copy number variations, CNV https://mc06.manuscriptcentral.com/bcb-pubs Page 1 of 354 Biochemistry and Cell Biology 1 Copy number variation in fetal alcohol spectrum disorder 2 Mehdi Zarrei,a Geoffrey G. Hicks,b James N. Reynolds,c,d Bhooma Thiruvahindrapuram,a 3 Worrawat Engchuan,a Molly Pind,b Sylvia Lamoureux,a John Wei,a Zhouzhi Wang,a Christian R. 4 Marshall,a Richard F. Wintle,a Albert E. Chudleye,f and Stephen W. Scherer,a,g 5 aThe Centre for Applied Genomics and Program in Genetics and Genome Biology, The Hospital 6 for Sick Children, Toronto, Ontario, Canada 7 bRegenerative Medicine Program, University of Manitoba, Winnipeg, Canada 8 cCentre for Neuroscience Studies, Queen's University, Kingston, Ontario, Canada. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
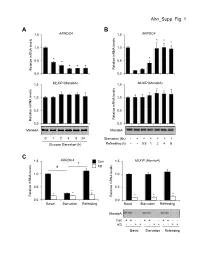
Ahn Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0
Ahn_Supp. Fig. 1 AB 1.5 ARRDC4 1.5 ARRDC4 * * * 1.0 1.0 * * 0.5 * 0.5 * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 1.5 MLXIP (MondoA) 1.5 MLXIP (MondoA) 1.0 1.0 0.5 0.5 Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative 0.0 0.0 MondoA MondoA 0124824 Starvation (6h) -++++++ Glucose Starvation (h) Refeeding (h) --0.51248 C 1.5 ARRDC4 1.5 MLXIP (MondoA) † Con # KD 1.0 1.0 0.5 0.5 * * * * Relative mRNA levels mRNA Relative Relative mRNA levels mRNA Relative * * 0.0 0.0 BasalStarvation Refeeding BasalStarvation Refeeding MondoA Con + + - - + + - - + + - - KD - - + + - - + + - - + + BasalStarvation Refeeding Supplemental Figure 1. Glucose-mediated regulation of ARRDC4 is dependent on MondoA in human skeletal myotubes. (A) (top) ARRDC4 and MLXIP (MondoA) mRNA levels were determined by qRT-PCR in human skeletal myotubes following deprivation of glucose at the indicated time (n=4). (bottom) Representative Western blot analysis of MondoA demonstrating the effect of glucose deprivation. *p<0.05 vs. 0h. (B) (top) ARRDC4 and MLXIP (MondoA) expression in human myotubes following a 6h glucose removal and refeeding at the times indicated (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs Starvation 6h. (C) (top) Expression of ARRDC4 and MLXIP in human myotubes following deprivation and refeeding of glucose in the absence or presence of siRNA-mediated MondoA KD (n=4). (bottom) Corresponding Western blot analysis. *p<0.05 vs siControl. # p<0.05. § p<0.05. The data represents mean ± SD. All statistical significance determined by one-way ANOVA with Tukey multiple comparison post-hoc test. -

Transcriptomic Characterisation of the Molecular Mechanisms Induced by Rgma During Skeletal Muscle Hyperplasia and Hypertrophy
Transcriptomic Characterisation of the Molecular Mechanisms Induced by RGMa During Skeletal Muscle Hyperplasia and Hypertrophy Aline Gonçalves Lio Copola Universidade Federal de Minas Gerais Íria Gabriela Dias dos Santos Universidade Federal de Minas Gerais Luiz Lehmann Coutinho Universidade de São Paulo Luiz Eduardo Vieira Del Bem Universidade Federal de Minas Gerais Paulo Henrique de Almeida Campos Junior Federal University of São João del-Rei Júlia Meireles Nogueira Universidade Federal de Minas Gerais Alinne do Carmo Costa Universidade Federal de Minas Gerais Gerluza Aparecida Borges Silva Universidade Federal de Minas Gerais Erika Cristina Jorge ( [email protected] ) Universidade Federal de Minas Gerais Research Article Keywords: Axon Guidance, myogenesis, hypertrophy, hyperplasia, skeletal muscle differentiation, transcriptomic analysis Posted Date: June 29th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-646954/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/25 Abstract Background: The repulsive guidance molecule a (RGMa) is a GPI-anchor axon guidance molecule rst found to play important roles during neuronal development. RGMa expression patterns and signalling pathways via Neogenin and/or as BMP coreceptors indicated that this axon guidance molecule could also be working in other processes and diseases, including during myogenesis. Previous works have consistently shown that RGMa is expressed in skeletal muscle cells and that its overexpression induces both nuclei accretion and hypertrophy in muscle cell lineages. However, the cellular components and molecular mechanisms induced by RGMa during the differentiation of skeletal muscle cells are poorly understood. In this work, the global transcription expression prole of RGMa-treated C2C12 myoblasts during the differentiation stage, obtained by RNA- seq, were reported. -

Characterization of the Macrophage Transcriptome in Glomerulonephritis-Susceptible and -Resistant Rat Strains
Genes and Immunity (2011) 12, 78–89 & 2011 Macmillan Publishers Limited All rights reserved 1466-4879/11 www.nature.com/gene ORIGINAL ARTICLE Characterization of the macrophage transcriptome in glomerulonephritis-susceptible and -resistant rat strains K Maratou1, J Behmoaras2, C Fewings1, P Srivastava1, Z D’Souza1, J Smith3, L Game4, T Cook2 and T Aitman1 1Physiological Genomics and Medicine Group, MRC Clinical Sciences Centre, Imperial College London, London, UK; 2Centre for Complement and Inflammation Research, Imperial College London, London, UK; 3Renal Section, Imperial College London, London, UK and 4Genomics Laboratory, MRC Clinical Sciences Centre, London, UK Crescentic glomerulonephritis (CRGN) is a major cause of rapidly progressive renal failure for which the underlying genetic basis is unknown. Wistar–Kyoto (WKY) rats show marked susceptibility to CRGN, whereas Lewis rats are resistant. Glomerular injury and crescent formation are macrophage dependent and mainly explained by seven quantitative trait loci (Crgn1–7). Here, we used microarray analysis in basal and lipopolysaccharide (LPS)-stimulated macrophages to identify genes that reside on pathways predisposing WKY rats to CRGN. We detected 97 novel positional candidates for the uncharacterized Crgn3–7. We identified 10 additional secondary effector genes with profound differences in expression between the two strains (45-fold change, o1% false discovery rate) for basal and LPS-stimulated macrophages. Moreover, we identified eight genes with differentially expressed alternatively spliced isoforms, by using an in-depth analysis at the probe level that allowed us to discard false positives owing to polymorphisms between the two rat strains. Pathway analysis identified several common linked pathways, enriched for differentially expressed genes, which affect macrophage activation. -

A High-Resolution Integrated Map of Copy Number Polymorphisms Within
Nicholas et al. BMC Genomics 2011, 12:414 http://www.biomedcentral.com/1471-2164/12/414 RESEARCHARTICLE Open Access A high-resolution integrated map of copy number polymorphisms within and between breeds of the modern domesticated dog Thomas J Nicholas1, Carl Baker1, Evan E Eichler1,2 and Joshua M Akey1* Abstract Background: Structural variation contributes to the rich genetic and phenotypic diversity of the modern domestic dog, Canis lupus familiaris, although compared to other organisms, catalogs of canine copy number variants (CNVs) are poorly defined. To this end, we developed a customized high-density tiling array across the canine genome and used it to discover CNVs in nine genetically diverse dogs and a gray wolf. Results: In total, we identified 403 CNVs that overlap 401 genes, which are enriched for defense/immunity, oxidoreductase, protease, receptor, signaling molecule and transporter genes. Furthermore, we performed detailed comparisons between CNVs located within versus outside of segmental duplications (SDs) and find that CNVs in SDs are enriched for gene content and complexity. Finally, we compiled all known dog CNV regions and genotyped them with a custom aCGH chip in 61 dogs from 12 diverse breeds. These data allowed us to perform the first population genetics analysis of canine structural variation and identify CNVs that potentially contribute to breed specific traits. Conclusions: Our comprehensive analysis of canine CNVs will be an important resource in genetically dissecting canine phenotypic and behavioral variation. Background domestication, examine relatedness among breeds, and The domestication of the modern dog from their wolf identify signatures of artificial selection [4,7-9].