The Cell Biology of Systemic Hyperinflammation Resulting from Failed Cytolytic Target Cell Killing
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genome-Wide Rnai Screens Identify Genes Required for Ricin and PE Intoxications
Developmental Cell Article Genome-Wide RNAi Screens Identify Genes Required for Ricin and PE Intoxications Dimitri Moreau,1 Pankaj Kumar,1 Shyi Chyi Wang,1 Alexandre Chaumet,1 Shin Yi Chew,1 He´ le` ne Chevalley,1 and Fre´ de´ ric Bard1,* 1Institute of Molecular and Cell Biology, 61 Biopolis Drive, Proteos, Singapore 138673, Singapore *Correspondence: [email protected] DOI 10.1016/j.devcel.2011.06.014 SUMMARY In the lumen of the ER, these toxins are thought to interact with elements of the ER-associated degradation (ERAD) pathway, Protein toxins such as Ricin and Pseudomonas which targets misfolded proteins in the ER for degradation. exotoxin (PE) pose major public health challenges. This interaction is proposed to allow translocation to the cytosol Both toxins depend on host cell machinery for inter- without resulting in toxin degradation (Johannes and Ro¨ mer, nalization, retrograde trafficking from endosomes 2010). to the ER, and translocation to cytosol. Although Obviously, this complex set of membrane-trafficking and both toxins follow a similar intracellular route, it is membrane-translocation events involves many host proteins, some of which have already been described (Johannes and unknown how much they rely on the same genes. Ro¨ mer, 2010; Sandvig et al., 2010). Altering the function of these Here we conducted two genome-wide RNAi screens host proteins could in theory provide a toxin antidote. identifying genes required for intoxication and Consistently, inhibition of retrograde traffic by drugs such as demonstrating that requirements are strikingly Brefeldin A (Sandvig et al., 1991)(Yoshida et al., 1991) or Golgi- different between PE and Ricin, with only 13% over- cide A (Sa´ enz et al., 2009) and Retro-1 and 2 (Stechmann et al., lap. -

Regular Article
From www.bloodjournal.org by guest on April 6, 2015. For personal use only. Regular Article IMMUNOBIOLOGY Hemophagocytic lymphohistiocytosis caused by dominant-negative mutations in STXBP2 that inhibit SNARE-mediated membrane fusion Waldo A. Spessott,1 Maria L. Sanmillan,1 Margaret E. McCormick,1 Nishant Patel,2 Joyce Villanueva,3 Kejian Zhang,4 Kim E. Nichols,5 and Claudio G. Giraudo1 1Department of Pathology and Laboratory Medicine, and 2Division of Oncology, Department of Pediatrics, The Children’s Hospital of Philadelphia, University of Pennsylvania, Philadelphia, PA; 3Division of Bone Marrow Transplant and Immune Deficiency, and 4Division of Human Genetics, Cincinnati Children’s Hospital Medical Center, Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH; and 5Division of Cancer Predisposition, Department of Oncology, St. Jude Children’s Research Hospital, Memphis, TN Key Points Familial hemophagocytic lymphohistiocytosis (F-HLH) and Griscelli syndrome type 2 (GS) are life-threatening immunodeficiencies characterized by impaired cytotoxic T lymphocyte • Monoallelic STXBP2 mutations (CTL) and natural killer (NK) cell lytic activity. In the majority of cases, these disorders are affecting codon 65 impair caused by biallelic inactivating germline mutations in genes such as RAB27A (GS) and PRF1, lymphocyte cytotoxicity and UNC13D, STX11,andSTXBP2 (F-HLH). Although monoallelic (ie, heterozygous) mutations contribute to hemophagocytic have been identified in certain patients, the clinical significance and molecular mechanisms lymphohistiocytosis. by which these mutations influence CTL and NK cell function remain poorly understood. • Munc18-2R65Q/W mutant Here, we characterize 2 novel monoallelic hemophagocytic lymphohistiocytosis (HLH)- associated mutations affecting codon 65 of STXPB2, the gene encoding Munc18-2, a member proteins function in a dominant- of the SEC/MUNC18 family. -

Clinical Spectrum Ofstx1b-Related Epileptic Disorders
ARTICLE OPEN ACCESS Clinical spectrum of STX1B-related epileptic disorders Stefan Wolking, MD, Patrick May, PhD, Davide Mei, PhD, Rikke S. Møller, PhD, Simona Balestrini, PhD, Correspondence Katherine L. Helbig, MS, Cecilia Desmettre Altuzarra, MD, Nicolas Chatron, PhD, Charu Kaiwar, MD, Dr. Lerche Katharina Stohr,¨ MD, Peter Widdess-Walsh, MB, Bryce A. Mendelsohn, PhD, Adam Numis, MD, holger.lerche@ Maria R. Cilio, PhD, Wim Van Paesschen, MD, Lene L. Svendsen, MD, Stephanie Oates, MD, Elaine Hughes, MD, uni-tuebingen.de Sushma Goyal, MD, Kathleen Brown, MS, Margarita Sifuentes Saenz, MD, Thomas Dorn, MD, Hiltrud Muhle, MD, Alistair T. Pagnamenta, PhD, Dimitris V. Vavoulis, PhD, Samantha J.L. Knight, PhD, Jenny C. Taylor, PhD, Maria Paola Canevini, MD, Francesca Darra, MD, Ralitza H. Gavrilova, MD, Zoe¨ Powis, MS, Shan Tang, PhD, Justus Marquetand, MD, Martin Armstrong, PhD, Duncan McHale, PhD, Eric W. Klee, PhD, Gerhard J. Kluger, MD, Daniel H. Lowenstein, MD, Sarah Weckhuysen, PhD, Deb K. Pal, PhD, Ingo Helbig, MD, Renzo Guerrini, MD, Rhys H. Thomas, PhD, Mark I. Rees, PhD, Gaetan Lesca, PhD, Sanjay M. Sisodiya, PhD, Yvonne G. Weber, MD, Dennis Lal, PhD, Carla Marini, PhD, Holger Lerche, MD, and Julian Schubert, PhD Neurology® 2019;92:e1238-e1249. doi:10.1212/WNL.0000000000007089 Abstract Objective The aim of this study was to expand the spectrum of epilepsy syndromes related to STX1B, encoding the presynaptic protein syntaxin-1B, and establish genotype-phenotype correlations by identifying further disease- related variants. Methods We used next-generation sequencing in the framework of research projects and diagnostic testing. Clinical data and EEGs were reviewed, including already published cases. -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -

Syntaxin Binding Mechanism and Disease-Causing Mutations In
Syntaxin binding mechanism and disease-causing PNAS PLUS mutations in Munc18-2 Yvonne Hackmanna, Stephen C. Grahama,1,2, Stephan Ehlb, Stefan Höningc, Kai Lehmbergd, Maurizio Aricòe, David J. Owena, and Gillian M. Griffithsa,2 aCambridge Institute for Medical Research, University of Cambridge Biomedical Campus, University of Cambridge, Cambridge CB2 0XY, United Kingdom; bCentre of Chronic Immunodeficiency, 79106 Freiburg, Germany; cInstitute for Biochemistry I and Center for Molecular Medicine Cologne, University of Cologne, 50931 Cologne, Germany; dDepartment of Paediatric Haematology and Oncology, University Medical Center Hamburg Eppendorf, 20246 Hamburg, Germany; and ePediatric Hematology Oncology Network, Istituto Toscana Tumori, 50139 Florence, Italy Edited* by Peter Cresswell, Yale University School of Medicine, New Haven, CT, and approved October 11, 2013 (received for review July 18, 2013) Mutations in either syntaxin 11 (Stx11) or Munc18-2 abolish Munc18-2 belongs to the Sec1/Munc18-like (SM) protein cytotoxic T lymphocytes (CTL) and natural killer cell (NK) cytotox- family, whose members are all ∼600 residues long and are in- icity, and give rise to familial hemophagocytic lymphohistiocytosis volved in regulation of SNARE-mediated membrane fusion (FHL4 or FHL5, respectively). Although Munc18-2 is known to events (9, 10). The two closest homologs of Munc18-2 are interact with Stx11, little is known about the molecular mecha- Munc18-1, which is crucial for neurotransmitter secretion in nisms governing the specificity of this interaction or how in vitro neurons (11, 12), and Munc18-3, which is more widely expressed IL-2 activation leads to compensation of CTL and NK cytotoxicity. and is involved in Glut4 translocation (13). -

Mapping Transmembrane Binding Partners for E-Cadherin Ectodomains
SUPPLEMENTARY INFORMATION TITLE: Mapping transmembrane binding partners for E-cadherin ectodomains. AUTHORS: Omer Shafraz 1, Bin Xie 2, Soichiro Yamada 1, Sanjeevi Sivasankar 1, 2, * AFFILIATION: 1 Department of Biomedical Engineering, 2 Biophysics Graduate Group, University of California, Davis, CA 95616. *CORRESPONDING AUTHOR: Sanjeevi Sivasankar, Tel: (530)-754-0840, Email: [email protected] Figure S1: Western blots a. EC-BioID, WT and Ecad-KO cell lysates stained for Ecad and tubulin. b. HRP-streptavidin staining of biotinylated proteins eluted from streptavidin coated magnetic beads incubated with cell lysates of EC-BioID with (+) and without (-) exogenous biotin. c. C-BioID, WT and Ecad-KO cell lysates stained for Ecad and tubulin. d. HRP-streptavidin staining of biotinylated proteins eluted from streptavidin coated magnetic beads incubated with cell lysates of C-BioID with (+) and without (-) exogenous biotin. (+) Biotin (-) Biotin Sample 1 Sample 2 Sample 3 Sample 4 Sample 1 Sample 2 Sample 3 Sample 4 Percent Percent Percent Percent Percent Percent Percent Percent Gene ID Coverage Coverage Coverage Coverage Coverage Coverage Coverage Coverage CDH1 29.6 31.4 41.1 36.5 10.8 6.7 28.8 29.1 DSG2 26 14.6 45 37 0.8 1.9 1.6 18.7 CXADR 30.2 26.2 32.7 27.1 0.0 0.0 0.0 6.9 EFNB1 24.3 30.6 24 30.3 0.0 0.0 0.0 0.0 ITGA2 16.5 22.2 30.1 33.4 1.1 1.1 5.2 7.2 CDH3 21.8 9.7 20.6 25.3 1.3 1.3 0.0 0.0 ITGB1 11.8 16.7 23.9 20.3 0.0 2.9 8.5 5.8 DSC3 9.7 7.5 11.5 13.3 0.0 0.0 2.6 0.0 EPHA2 23.2 31.6 31.6 30.5 0.8 0.0 0.0 5.7 ITGB4 21.8 27.8 33.1 30.7 0.0 1.2 3.9 4.4 ITGB3 23.5 22.2 26.8 24.7 0.0 0.0 5.2 9.1 CDH6 22.8 18.1 28.6 24.3 0.0 0.0 0.0 9.1 CDH17 8.8 12.4 20.7 18.4 0.0 0.0 0.0 0.0 ITGB6 12.7 10.4 14 17.1 0.0 0.0 0.0 1.7 EPHB4 11.4 8.1 14.2 16.3 0.0 0.0 0.0 0.0 ITGB8 5 10 15 17.6 0.0 0.0 0.0 0.0 ITGB5 6.2 9.5 15.2 13.8 0.0 0.0 0.0 0.0 EPHB2 8.5 4.8 9.8 12.1 0.0 0.0 0.0 0.0 CDH24 5.9 7.2 8.3 9 0.0 0.0 0.0 0.0 Table S1: EC-BioID transmembrane protein hits. -

Regulation Pathway of Mesenchymal Stem Cell Immune Dendritic Cell
Downloaded from http://www.jimmunol.org/ by guest on September 26, 2021 is online at: average * The Journal of Immunology , 13 of which you can access for free at: 2010; 185:5102-5110; Prepublished online 1 from submission to initial decision 4 weeks from acceptance to publication October 2010; doi: 10.4049/jimmunol.1001332 http://www.jimmunol.org/content/185/9/5102 Inhibition of Immune Synapse by Altered Dendritic Cell Actin Distribution: A New Pathway of Mesenchymal Stem Cell Immune Regulation Alessandra Aldinucci, Lisa Rizzetto, Laura Pieri, Daniele Nosi, Paolo Romagnoli, Tiziana Biagioli, Benedetta Mazzanti, Riccardo Saccardi, Luca Beltrame, Luca Massacesi, Duccio Cavalieri and Clara Ballerini J Immunol cites 38 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2010/10/01/jimmunol.100133 2.DC1 This article http://www.jimmunol.org/content/185/9/5102.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2010 by The American Association of -
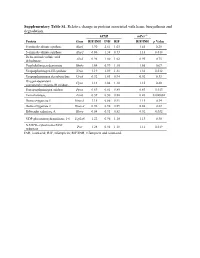
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator- -

The Biogenesis of Lysosomes and Lysosome-Related Organelles
Downloaded from http://cshperspectives.cshlp.org/ on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press The Biogenesis of Lysosomes and Lysosome-Related Organelles J. Paul Luzio1, Yvonne Hackmann2, Nele M.G. Dieckmann2, and Gillian M. Griffiths2 1Department of Clinical Biochemistry, Cambridge Institute for Medical Research, University of Cambridge, Cambridge Biomedical Campus, Cambridge CB2 0XY, United Kingdom 2Department of Medicine, Cambridge Institute for Medical Research, University of Cambridge, Cambridge Biomedical Campus, Cambridge CB2 0XY, United Kingdom Correspondence: [email protected] Lysosomes were once considered the end point of endocytosis, simply used for macromol- ecule degradation. They are now recognized to be dynamic organelles, able to fuse with a variety of targets and to be re-formed after fusion events. They are also now known to be the site of nutrient sensing and signaling to the cell nucleus. In addition, lysosomes are secretory organelles, with specialized machinery for regulated secretion of proteins in some cell types. The biogenesis of lysosomes and lysosome-related organelles is discussed, taking into account their dynamic nature and multiple roles. WHAT IS A LYSOSOME? content and sometimes multilamellar mem- brane whorls (see Klumperman and Raposo ysosomes are membrane-bound organelles 2014). Lysosomes are distinguished from late Lcontaining more than 50 acid hydrolases endosomes by the absence of mannose-6-phos- that function in the degradation of macromol- phate receptors (MPRs) (Brown et -

Integrated Bioinformatics Analysis Reveals Novel Key Biomarkers and Potential Candidate Small Molecule Drugs in Gestational Diabetes Mellitus
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.09.434569; this version posted March 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Integrated bioinformatics analysis reveals novel key biomarkers and potential candidate small molecule drugs in gestational diabetes mellitus Basavaraj Vastrad1, Chanabasayya Vastrad*2, Anandkumar Tengli3 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karnataka, India. 3. Department of Pharmaceutical Chemistry, JSS College of Pharmacy, Mysuru and JSS Academy of Higher Education & Research, Mysuru, Karnataka, 570015, India * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2021.03.09.434569; this version posted March 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Gestational diabetes mellitus (GDM) is one of the metabolic diseases during pregnancy. The identification of the central molecular mechanisms liable for the disease pathogenesis might lead to the advancement of new therapeutic options. The current investigation aimed to identify central differentially expressed genes (DEGs) in GDM. The transcription profiling by array data (E-MTAB-6418) was obtained from the ArrayExpress database. The DEGs between GDM samples and non GDM samples were analyzed with limma package. Gene ontology (GO) and REACTOME enrichment analysis were performed using ToppGene. Then we constructed the protein-protein interaction (PPI) network of DEGs by the Search Tool for the Retrieval of Interacting Genes database (STRING) and module analysis was performed. -

Dynamic Palmitoylation Events Following T-Cell Receptor Signaling
bioRxiv preprint doi: https://doi.org/10.1101/831388; this version posted November 5, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Dynamic Palmitoylation Events Following T-Cell Receptor Signaling Eliot Morrison1, Tatjana Wegner1, Andres Ernesto Zucchetti2, Miguel Álvaro-Benito1, Ashley Zheng1, 3 4 5 2 1* Stefanie Kliche , Eberhard Krause , Britta Brügger , Claire Hivroz & Christian Freund 1Freie Universität Berlin, Institute for Chemistry & Biochemistry, Laboratory of Protein Biochemistry, Thielallee 63, 14195 Berlin 2Institut Curie, PSL Research University, INSERM U932, Integrative analysis of T cell activation team, 26 rue d'Ulm, 75248, Paris Cedex 05, France 3Otto-von-Guericke-University, Institute of Molecular and Clinical Immunology, Health Campus Immunology, Infectiology and Inflammation, Leipziger Strasse 44, 39120 Magdeburg, Germany 4Leibniz Institute for Molecular Pharmacology, Mass Spectrometry Unit, Robert-Rössle-Str 10, 13125 Berlin 5Heidelberg University Biochemistry Center (BZH), Im Neuenheimer Feld 328, 69120, Heidelberg, Germany. *Corresponding address: [email protected] Abstract Palmitoylation is the reversible addition of palmitate to cysteine via a thioester linkage. Following stimulation of the T-cell receptor we find a number of proteins are newly palmitoylated, including those involved in vesicle- mediated transport and Ras signal transduction. Among these stimulation-dependent palmitoylation targets are the v-SNARE VAMP7, important for docking of vesicular LAT during TCR signaling, and the largely undescribed palmitoyl acyltransferase DHHC18 that is expressed in two isoforms in T cells. Using our newly developed On- Plate Palmitoylation Assay (OPPA), we show DHHC18 is capable of palmitoylating VAMP7 at Cys183. -

STX11 Functions As a Novel Tumor Suppressor Gene in Peripheral T&
STX11 functions as a novel tumor suppressor gene in peripheral T-cell lymphomas Noriaki Yoshida,1,2,3 Shinobu Tsuzuki,1 Kennosuke Karube,1,8 Taishi Takahara,1,4 Miyuki Suguro,1 Hiroaki Miyoshi,3 Momoko Nishikori,5 Masanori Shimoyama,6 Kunihiro Tsukasaki,7 Koichi Ohshima3 and Masao Seto1,2,3 1Division of Molecular Medicine, Aichi Cancer Center Research Institute, Nagoya; 2Department of Cancer Genetics, Nagoya University Graduate School of Medicine at Aichi Cancer Center Research Institute, Nagoya; 3Department of Pathology, Kurume University School of Medicine, Kurume; 4Department of Pathology and Laboratory Medicine, Nagoya University Graduate School of Medicine, Nagoya; 5Department of Hematology and Oncology, Graduate School of Medicine, Kyoto University, Kyoto; 6Multi-institutional Clinical Trial Support Center, National Cancer Center, Tokyo; 7Department of Hematology, National Cancer Center Hospital East, Kashiwa, Japan; Key words Peripheral T-cell lymphomas (PTCL) are a heterogeneous group of non-Hodgkin Functional analyses, genomic loss of 6q24, peripheral T- lymphomas with poor prognosis. Their molecular pathogenesis has not been cell lymphomas, STX11, tumor suppressor gene entirely elucidated. We previously showed that 6q24 is one of the most fre- Correspondence quently deleted regions in primary thyroid T-cell lymphoma. In this study, we Masao Seto, Department of Pathology, Kurume University extended the analysis to other subtypes of PTCL and performed functional assays School of Medicine, 67 Asahimachi, Kurume, Fukuoka to identify the causative genes of PTCL that are located on 6q24. Genomic loss of 830-0011, Japan. 6q24 was observed in 14 of 232 (6%) PTCL cases. The genomic loss regions identi- Tel: +81-942-35-3311 (Ext.