Ublituximab (UTX), a Novel Glycoengineered Anti-CD20 Monoclonal Antibody (Mab), in Patients with Relapsing Forms of Multiple Sclerosis (RMS)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Predictive QSAR Tools to Aid in Early Process Development of Monoclonal Antibodies
Predictive QSAR tools to aid in early process development of monoclonal antibodies John Micael Andreas Karlberg Published work submitted to Newcastle University for the degree of Doctor of Philosophy in the School of Engineering November 2019 Abstract Monoclonal antibodies (mAbs) have become one of the fastest growing markets for diagnostic and therapeutic treatments over the last 30 years with a global sales revenue around $89 billion reported in 2017. A popular framework widely used in pharmaceutical industries for designing manufacturing processes for mAbs is Quality by Design (QbD) due to providing a structured and systematic approach in investigation and screening process parameters that might influence the product quality. However, due to the large number of product quality attributes (CQAs) and process parameters that exist in an mAb process platform, extensive investigation is needed to characterise their impact on the product quality which makes the process development costly and time consuming. There is thus an urgent need for methods and tools that can be used for early risk-based selection of critical product properties and process factors to reduce the number of potential factors that have to be investigated, thereby aiding in speeding up the process development and reduce costs. In this study, a framework for predictive model development based on Quantitative Structure- Activity Relationship (QSAR) modelling was developed to link structural features and properties of mAbs to Hydrophobic Interaction Chromatography (HIC) retention times and expressed mAb yield from HEK cells. Model development was based on a structured approach for incremental model refinement and evaluation that aided in increasing model performance until becoming acceptable in accordance to the OECD guidelines for QSAR models. -

Ublituximab (TG-1101) in Combination with Umbralisib (TGR-1202) for Chronic Lymphocytic Leukaemia NIHRIO (HSRIC) ID: 12230 NICE ID: 9540
NIHR Innovation Observatory Evidence Briefing: SEPTEMBER 2017 Ublituximab (TG-1101) in combination with umbralisib (TGR-1202) for chronic lymphocytic leukaemia NIHRIO (HSRIC) ID: 12230 NICE ID: 9540 LAY SUMMARY Chronic lymphocytic leukaemia is a type of blood cancer that develops inside the bone marrows. This produces abnormal types of white blood cells (lymphocytes) which do not function properly, hence, do not protect from infections the way they should do. Chronic types of leukaemia develop slowly over a long time. Symptoms of this cancer usually only develop at a later stage of the disease. They may include frequent infections, persistent tiredness, shortness of breath, pale skin, bleeding and bruising more easily and others. The cause of chronic lymphocytic leukaemia is not yet fully understood but risk factors may include age (older people), exposure to chemicals, family history, gender or ethnicity. Ublituximab in combination with umbralisib is under development for the treatment of chronic lymphocytic leukaemia and other types of blood cancers. They both act in different ways to suppress the production and growth of the abnormal lymphocytes caused by bindings to certain cells and proteins in the body that produces the cancer. If licensed, it will offer an additional treatment option for patients with chronic lymphocytic leukaemia. This briefing is based on information available at the time of research and a limited literature search. It is not intended to be a definitive statement on the safety, efficacy or effectiveness of the health technology covered and should not be used for commercial purposes or commissioning without additional information. This briefing presents independent research funded by the National Institute for Health Research (NIHR). -

Tanibirumab (CUI C3490677) Add to Cart
5/17/2018 NCI Metathesaurus Contains Exact Match Begins With Name Code Property Relationship Source ALL Advanced Search NCIm Version: 201706 Version 2.8 (using LexEVS 6.5) Home | NCIt Hierarchy | Sources | Help Suggest changes to this concept Tanibirumab (CUI C3490677) Add to Cart Table of Contents Terms & Properties Synonym Details Relationships By Source Terms & Properties Concept Unique Identifier (CUI): C3490677 NCI Thesaurus Code: C102877 (see NCI Thesaurus info) Semantic Type: Immunologic Factor Semantic Type: Amino Acid, Peptide, or Protein Semantic Type: Pharmacologic Substance NCIt Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor tyrosine kinase expressed by endothelial cells, while VEGF is overexpressed in many tumors and is correlated to tumor progression. PDQ Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor -

The Future of Antibodies As Cancer Drugs
REVIEWS Drug Discovery Today Volume 17, Numbers 17/18 September 2012 The biopharmaceutical industry’s pipeline of anticancer antibodies includes 165 candidates with substantial diversity in composition, targets and mechanisms of action that hold promise to be the cancer drugs of the future. Reviews FOUNDATION REVIEW Foundation review: The future of antibodies as cancer drugs 1 2 Dr Janice Reichert Janice M. Reichert and Eugen Dhimolea is Research Assistant Professor at Tufts 1 Center for the Study of Drug Development, Tufts University School of Medicine, 75 Kneeland Street, University’s Center for the Study of Drug Development Suite 1100, Boston, MA 02111, USA 2 (CSDD). She is also Founder Department of Medical Oncology, Dana-Farber Cancer Institute/Harvard Medical School, 77 Louis Pasteur Ave., and Editor-in-Chief of mAbs, Harvard Institutes of Medicine, Room 309, Boston, MA 02215, USA a peer-reviewed, PubMed- indexed biomedical journal that focuses on topics relevant to antibody research Targeted therapeutics such as monoclonal antibodies (mAbs) have proven and development; President of the board of directors of The Antibody Society; and a member of the board successful as cancer drugs. To profile products that could be marketed in of the Peptide Therapeutics Foundation. At CSDD, the future, we examined the current commercial clinical pipeline of mAb Dr Reichert studies innovation in the pharmaceutical and biotechnology industries. Her work focuses on candidates for cancer. Our analysis revealed trends toward development of strategic analyses of investigational candidates and marketed products, with an emphasis on the clinical a variety of noncanonical mAbs, including antibody–drug conjugates development and approval of new therapeutics and (ADCs), bispecific antibodies, engineered antibodies and antibody vaccines. -

2017 Immuno-Oncology Medicines in Development
2017 Immuno-Oncology Medicines in Development Adoptive Cell Therapies Drug Name Organization Indication Development Phase ACTR087 + rituximab Unum Therapeutics B-cell lymphoma Phase I (antibody-coupled T-cell receptor Cambridge, MA www.unumrx.com immunotherapy + rituximab) AFP TCR Adaptimmune liver Phase I (T-cell receptor cell therapy) Philadelphia, PA www.adaptimmune.com anti-BCMA CAR-T cell therapy Juno Therapeutics multiple myeloma Phase I Seattle, WA www.junotherapeutics.com Memorial Sloan Kettering New York, NY anti-CD19 "armored" CAR-T Juno Therapeutics recurrent/relapsed chronic Phase I cell therapy Seattle, WA lymphocytic leukemia (CLL) www.junotherapeutics.com Memorial Sloan Kettering New York, NY anti-CD19 CAR-T cell therapy Intrexon B-cell malignancies Phase I Germantown, MD www.dna.com ZIOPHARM Oncology www.ziopharm.com Boston, MA anti-CD19 CAR-T cell therapy Kite Pharma hematological malignancies Phase I (second generation) Santa Monica, CA www.kitepharma.com National Cancer Institute Bethesda, MD Medicines in Development: Immuno-Oncology 1 Adoptive Cell Therapies Drug Name Organization Indication Development Phase anti-CEA CAR-T therapy Sorrento Therapeutics liver metastases Phase I San Diego, CA www.sorrentotherapeutics.com TNK Therapeutics San Diego, CA anti-PSMA CAR-T cell therapy TNK Therapeutics cancer Phase I San Diego, CA www.sorrentotherapeutics.com Sorrento Therapeutics San Diego, CA ATA520 Atara Biotherapeutics multiple myeloma, Phase I (WT1-specific T lymphocyte South San Francisco, CA plasma cell leukemia www.atarabio.com -

Looking for Therapeutic Antibodies in Next Generation Sequencing Repositories
bioRxiv preprint doi: https://doi.org/10.1101/572958; this version posted March 10, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Title: Looking for Therapeutic Antibodies in Next Generation Sequencing Repositories. Authors: Konrad Krawczyk1*, Matthew Raybould2, Aleksandr Kovaltsuk2, Charlotte M. Deane2 1 NaturalAntibody, Hamburg, Germany 2 Oxford University Department of Statistics, Oxford, UK *Correspondence to [email protected] Abstract: Recently it has become possible to query the great diversity of natural antibody repertoires using Next Generation Sequencing (NGS). These methods are capable of producing millions of sequences in a single experiment. Here we compare Clinical Stage Therapeutic antibodies to the ~1b sequences from 60 independent sequencing studies in the Observed Antibody Space Database. Of the 242 post Phase I antibodies, we find 16 with sequence identity matches of 95% or better for both heavy and light chains. There are also 54 perfect matches to therapeutic CDR-H3 regions in the NGS outputs, suggesting a nontrivial amount of convergence between naturally observed sequences and those developed artificially. This has potential implications for both the discovery of antibody therapeutics and the legal protection of commercial antibodies. Introduction Antibodies are proteins in jawed vertebrates that recognize noxious molecules (antigens) for elimination. An organism expresses millions of diverse antibodies to increase the chances that some of them will be able to bind the foreign antigen, initiating the adaptive immune response. -

The Two Tontti Tudiul Lui Hi Ha Unit
THETWO TONTTI USTUDIUL 20170267753A1 LUI HI HA UNIT ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2017 /0267753 A1 Ehrenpreis (43 ) Pub . Date : Sep . 21 , 2017 ( 54 ) COMBINATION THERAPY FOR (52 ) U .S . CI. CO - ADMINISTRATION OF MONOCLONAL CPC .. .. CO7K 16 / 241 ( 2013 .01 ) ; A61K 39 / 3955 ANTIBODIES ( 2013 .01 ) ; A61K 31 /4706 ( 2013 .01 ) ; A61K 31 / 165 ( 2013 .01 ) ; CO7K 2317 /21 (2013 . 01 ) ; (71 ) Applicant: Eli D Ehrenpreis , Skokie , IL (US ) CO7K 2317/ 24 ( 2013. 01 ) ; A61K 2039/ 505 ( 2013 .01 ) (72 ) Inventor : Eli D Ehrenpreis, Skokie , IL (US ) (57 ) ABSTRACT Disclosed are methods for enhancing the efficacy of mono (21 ) Appl. No. : 15 /605 ,212 clonal antibody therapy , which entails co - administering a therapeutic monoclonal antibody , or a functional fragment (22 ) Filed : May 25 , 2017 thereof, and an effective amount of colchicine or hydroxy chloroquine , or a combination thereof, to a patient in need Related U . S . Application Data thereof . Also disclosed are methods of prolonging or increasing the time a monoclonal antibody remains in the (63 ) Continuation - in - part of application No . 14 / 947 , 193 , circulation of a patient, which entails co - administering a filed on Nov. 20 , 2015 . therapeutic monoclonal antibody , or a functional fragment ( 60 ) Provisional application No . 62/ 082, 682 , filed on Nov . of the monoclonal antibody , and an effective amount of 21 , 2014 . colchicine or hydroxychloroquine , or a combination thereof, to a patient in need thereof, wherein the time themonoclonal antibody remains in the circulation ( e . g . , blood serum ) of the Publication Classification patient is increased relative to the same regimen of admin (51 ) Int . -

Ep 3178848 A1
(19) TZZ¥__T (11) EP 3 178 848 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 14.06.2017 Bulletin 2017/24 C07K 16/28 (2006.01) A61K 39/395 (2006.01) C07K 16/30 (2006.01) (21) Application number: 15198715.3 (22) Date of filing: 09.12.2015 (84) Designated Contracting States: (72) Inventor: The designation of the inventor has not AL AT BE BG CH CY CZ DE DK EE ES FI FR GB yet been filed GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR (74) Representative: Cueni, Leah Noëmi et al Designated Extension States: F. Hoffmann-La Roche AG BA ME Patent Department Designated Validation States: Grenzacherstrasse 124 MA MD 4070 Basel (CH) (71) Applicant: F. Hoffmann-La Roche AG 4070 Basel (CH) (54) TYPE II ANTI-CD20 ANTIBODY FOR REDUCING FORMATION OF ANTI-DRUG ANTIBODIES (57) The present invention relates to methods of treating a disease, and methods for reduction of the formation of anti-drug antibodies (ADAs) in response to the administration of a therapeutic agent comprising administration of a Type II anti-CD20 antibody, e.g. obinutuzumab, to the subject prior to administration of the therapeutic agent. EP 3 178 848 A1 Printed by Jouve, 75001 PARIS (FR) EP 3 178 848 A1 Description Field of the Invention 5 [0001] The present invention relates to methods of treating a disease, and methods for reduction of the formation of anti-drug antibodies (ADAs) in response to the administration of a therapeutic agent. -
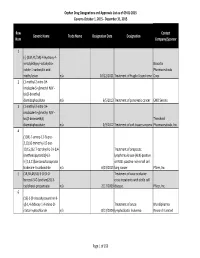
Orphan Drug Designation List
Orphan Drug Designations and Approvals List as of 09‐01‐2015 Governs October 1, 2015 ‐ December 31, 2015 Row Contact Generic Name Trade Name Designation Date Designation Num Company/Sponsor 1 (‐)‐(3aR,4S,7aR)‐4‐Hydroxy‐4‐ m‐tolylethynyl‐octahydro‐ Novartis indole‐1‐carboxylic acid Pharmaceuticals methyl ester n/a 10/12/2011 Treatment of Fragile X syndrome Corp. 2 (1‐methyl‐2‐nitro‐1H‐ imidazole‐5‐yl)methyl N,N'‐ bis(2‐broethyl) diamidophosphate n/a 6/5/2013 Treatment of pancreatic cancer EMD Serono 3 (1‐methyl‐2‐nitro‐1H‐ imidazole‐5‐yl)methyl N,N'‐ bis(2‐bromoethyl) Threshold diamidophosphate n/a 3/9/2012 Treatment of soft tissue sarcoma Pharmaceuticals, Inc. 4 (1OR)‐7‐amino‐12‐fluoro‐ 2,10,16‐trimethyl‐15 oxo‐ 10,15,16,17‐tetrahydro‐2H‐8,4‐ Treatment of anaplastic (metheno)pyrazolo[4,3‐ lymphoma kinase (ALK)‐positive h][2,5,11]benzoxadiazacyclote or ROS1‐positive non‐small cell tradecine‐3‐carbonitrile n/a 6/23/2015 lung cancer Pfizer, Inc. 5 (1R,3R,4R,5S)‐3‐O‐[2‐O‐ Treatment of vaso‐occlusive benzoyl‐3‐O‐(sodium(2S)‐3‐ crisis in patients with sickle cell cyclohexyl‐propanoate‐ n/a 2/17/2009 disease. Pfizer, Inc. 6 (1S)‐1‐(9‐deazahypoxanthin‐9‐ yl)‐1,4‐dideoxy‐1,4‐imino‐D‐ Treatment of acute Mundipharma ribitol‐hydrochloride n/a 8/13/2004 lymphoblastic leukemia Research Limited Page 1 of 359 Orphan Drug Designations and Approvals List as of 09‐01‐2015 Governs October 1, 2015 ‐ December 31, 2015 Row Contact Generic Name Trade Name Designation Date Designation Num Company/Sponsor 7 Treatment of chronic lymphocytic leukemia and related leukemias to include (1S)‐1‐(9‐deazahypoxanthin‐9‐ prolymphocytic leukemia, adult T‐ yl)‐1,4‐dideoxy‐1,4‐imino‐D‐ cell leukemia, and hairy cell Mundipharma ribitol‐hydrochloride n/a 8/10/2004 leukemia Research Ltd. -

Ep 3321281 A1
(19) TZZ¥¥ _ __T (11) EP 3 321 281 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 16.05.2018 Bulletin 2018/20 C07K 14/79 (2006.01) A61K 38/40 (2006.01) A61K 38/00 (2006.01) A61K 38/17 (2006.01) (2006.01) (2006.01) (21) Application number: 17192980.5 A61K 39/395 A61K 39/44 C07K 16/18 (2006.01) (22) Date of filing: 03.08.2012 (84) Designated Contracting States: • TIAN, Mei Mei AL AT BE BG CH CY CZ DE DK EE ES FI FR GB Coquitlam, BC V3J 7E6 (CA) GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO • VITALIS, Timothy PL PT RO RS SE SI SK SM TR Vancouver, BC V6Z 2N1 (CA) (30) Priority: 05.08.2011 US 201161515792 P (74) Representative: Gowshall, Jonathan Vallance Forresters IP LLP (62) Document number(s) of the earlier application(s) in Skygarden accordance with Art. 76 EPC: Erika-Mann-Strasse 11 12746240.6 / 2 739 649 80636 München (DE) (71) Applicant: biOasis Technologies Inc Remarks: Richmond BC V6X 2W8 (CA) •This application was filed on 25.09.2017 as a divisional application to the application mentioned (72) Inventors: under INID code 62. • JEFFERIES, Wilfred •Claims filed after the date of receipt of the divisional South Surrey, BC V4A 2V5 (CA) application (Rule 68(4) EPC). (54) P97 FRAGMENTS WITH TRANSFER ACTIVITY (57) The present invention is related to fragments of duction of the melanotransferrin fragment conjugated to human melanotransferrin (p97). In particular, this inven- a therapeutic or diagnostic agent to a subject. -

(12) STANDARD PATENT (11) Application No. AU 2012328524 B2 (19) AUSTRALIAN PATENT OFFICE
(12) STANDARD PATENT (11) Application No. AU 2012328524 B2 (19) AUSTRALIAN PATENT OFFICE (54) Title Protein formulations containing amino acids (51) International Patent Classification(s) A61K 47/16 (2006.01) A61K 39/395 (2006.01) A61K 9/08 (2006.01) A61K 47/12 (2006.0 1) (21) Application No: 2012328524 (22) Date of Filing: 2012.10.26 (87) WIPO No: W013/063510 (30) Priority Data (31) Number (32) Date (33) Country 61/552,688 2011.10.28 US (43) Publication Date: 2013.05.02 (44) Accepted Journal Date: 2017.05.18 (71) Applicant(s) Excelse Bio, Inc. (72) Inventor(s) Chang, Byeong Seon (74) Agent / Attorney Pizzeys Patent and Trade Mark Attorneys Pty Ltd, GPO Box 1374, BRISBANE, QLD, 4001, AU (56) Related Art Krishnan et al, Chapter 16; Formulation and Process Development Strategies for Manufacturing Biopharmaceuticals, Ed. Jameel & Hershenson, John Wiley & Sons, Inc., ISBN: 978-0-470-11812-2 Gokarn et al, Chapter 17: "Excipients for Protein Drugs"; Excipient Development for Pharmaceutical, Biotechnology, and Drug Delivery Systems, Ed. Katdare & Chaubal, Informa Healthcare USA, Inc., ISBN: 978-0-8493-2706-3 Falconer et al, Journal of Chemical Technology and Biotechnology (2011), Vol. 86, Pages 942-948 US 2009/0291076 Al WO 2011/109365 A2 US 2007/0116700 Al WO 2011/104381 A2 US 2007/0053900 Al Bolli et al, Biologicals (2010), Vol. 38, Pages 150-157 Maeder et al, Biologicals (2011), Vol. 39, Pages 43-49 Kamerzell et al, Advanced Drug Delivery Reviews (2011), Vol. 63, Pages 1118-1159 He et al, Journal of Pharmaceutical Sciences (2011), Vol. 100, -

Current Immunotherapy Approaches in Non-Hodgkin Lymphomas
Review Current Immunotherapy Approaches in Non-Hodgkin Lymphomas Robert Pytlik 1,2, Kamila Polgarova 2, Jana Karolova 2,3 and Pavel Klener 2,3,* 1 Institute of Haematology and Blood Transfusion, 128 00 Prague, Czech Republic; [email protected] 2 First Department of Internal Medicine-Hematology, University General Hospital in Prague and First Faculty of Medicine, Charles University, U Nemocnice 2, 128 08 Prague 2, 110 00 Prague, Czech Republic; [email protected] (K.P.); [email protected] (J.K.) 3 Institute of Pathological Physiology, First Faculty of Medicine, Charles University, 128 53 Prague, Czech Republic * Correspondence: [email protected]; Tel.: +420-224965933 Received: 30 October 2020; Accepted: 24 November 2020; Published: 27 November 2020 Abstract: Non-Hodgkin lymphomas (NHLs) are lymphoid malignancies of B- or T-cell origin. Despite great advances in treatment options and significant improvement of survival parameters, a large part of NHL patients either present with a chemotherapy-refractory disease or experience lymphoma relapse. Chemotherapy-based salvage therapy of relapsed/refractory NHL is, however, capable of re-inducing long-term remissions only in a minority of patients. Immunotherapy-based approaches, including bispecific antibodies, immune checkpoint inhibitors and genetically engineered T-cells carrying chimeric antigen receptors, single-agent or in combination with therapeutic monoclonal antibodies, immunomodulatory agents, chemotherapy or targeted agents demonstrated unprecedented clinical activity in heavily-pretreated patients with NHL, including chemotherapy-refractory cases with complex karyotype changes and other adverse prognostic factors. In this review, we recapitulate currently used immunotherapy modalities in NHL and discuss future perspectives of combinatorial immunotherapy strategies, including patient-tailored approaches.