Two-Armed Silicon
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Chapter 13.Pptx
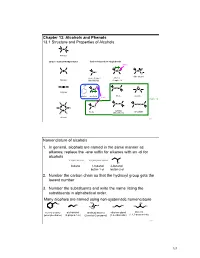
Chapter 13: Alcohols and Phenols 13.1 Structure and Properties of Alcohols C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic O C C C N C N C X O nitro alkane X= F, Cl, Br, I amines Alkenes Alkyl Halide Chapter 23 OH C C H O C O C C O C C Alkynes phenol alcohols ethers epoxide acidic Chapter 14 H H H C S C C C C S S C C S C C H C C sulfides thiols disulfide H H (thioethers) Arenes 253 Nomenclature of alcohols 1. In general, alcohols are named in the same manner as alkanes; replace the -ane suffix for alkanes with an -ol for alcohols CH3CH2CH2CH3 CH3CH2CH2CH2OH OH butane 1-butanol 2-butanol butan-1-ol butan-2-ol 2. Number the carbon chain so that the hydroxyl group gets the lowest number 3. Number the substituents and write the name listing the substituents in alphabetical order. Many alcohols are named using non-systematic nomenclature H C OH 3 OH OH C OH OH HO OH H3C HO H3C benzyl alcohol allyl alcohol tert-butyl alcohol ethylene glycol glycerol (phenylmethanol) (2-propen-1-ol) (2-methyl-2-propanol) (1,2-ethanediol) (1,2,3-propanetriol) 254 127 Alcohols are classified according to the H R C OH C OH H H degree of substitution of the carbon bearing H H 1° carbon the -OH group methanol primary alcohol primary (1°) : one alkyl substituent R R C OH C OH R R secondary (2°) : two alkyl substituents H R 2° carbon 3° carbon tertiary (3°) : three alkyl substituents secondary alcohol tertiary alcohol Physical properties of alcohols – the C-OH bond of alcohols has a significant dipole moment. -

Radical Approaches to Alangium and Mitragyna Alkaloids
Radical Approaches to Alangium and Mitragyna Alkaloids A Thesis Submitted for a PhD University of York Department of Chemistry 2010 Matthew James Palframan Abstract The work presented in this thesis has focused on the development of novel and concise syntheses of Alangium and Mitragyna alkaloids, and especial approaches towards (±)-protoemetinol (a), which is a key precursor of a range of Alangium alkaloids such as psychotrine (b) and deoxytubulosine (c). The approaches include the use of a key radical cyclisation to form the tri-cyclic core. O O O N N N O O O H H H H H H O N NH N Protoemetinol OH HO a Psychotrine Deoxytubulosine b c Chapter 1 gives a general overview of radical chemistry and it focuses on the application of radical intermolecular and intramolecular reactions in synthesis. Consideration is given to the mediator of radical reactions from the classic organotin reagents, to more recently developed alternative hydrides. An overview of previous synthetic approaches to a range of Alangium and Mitragyna alkaloids is then explored. Chapter 2 follows on from previous work within our group, involving the use of phosphorus hydride radical addition reactions, to alkenes or dienes, followed by a subsequent Horner-Wadsworth-Emmons reaction. It was expected that the tri-cyclic core of the Alangium alkaloids could be prepared by cyclisation of a 1,7-diene, using a phosphorus hydride to afford the phosphonate or phosphonothioate, however this approach was unsuccessful and it highlighted some limitations of the methodology. Chapter 3 explores the radical and ionic chemistry of a range of silanes. -

Annual Report 2015 Scanning Electron Microscopy Samples Undergoing Preparation at the Centre for Microscopy and Microanalysis Facility at AIBN
AIBNAustralian Institute for Bioengineering and Nanotechnology Annual Report 2015 Scanning electron microscopy samples undergoing preparation at the Centre for Microscopy and Microanalysis facility at AIBN. 2 - AIBN Annual Report 2015 AIBN Annual Report 2015 Vice-Chancellor and President’s Message 2 Discoveries and collaborations 28 MERS antibody building on Hendra virus work 30 Director’s Message 3 Cheaper, cleaner production of carbon fibre 31 AIBN Board 4 Getting biomarkers out of the skin Scientific Advisory Committee 5 with Micropatches 32 AIBN Research 6 High performance cluster boosts Alexandrov Group research capabilities 33 Synthetic biology drives diagnostic care 8 Self-assembled nanocapsules deliver results 34 Bernhardt Group Using theoretical and computational science Fast, cheap and easy to use for nanomaterials and fluids 9 tuberculosis screening 35 Cooper-White Group IAP removes barriers between academia Biomaterials development and discovery for and industry 36 regenerative medicine 10 AIBN Early-/Mid-Career Support Program 37 Gray Group Funding and recognition 38 Mammalian cell expression and bulk stem cell Australian Research Council funding 40 cultivation 11 National Health and Medical Research Council 41 Halley Group Biopolymers and starch for a Fellowships fuel research opportunities 42 cheaper, cleaner future 12 Awards highlight scientific excellence 43 Kendall Group Newly awarded research funding Gene and drug delivery and diagnostics commencing in 2015 44 through the skin 13 Facilities and infrastructure 46 Mahler -

Reductions and Reducing Agents
REDUCTIONS AND REDUCING AGENTS 1 Reductions and Reducing Agents • Basic definition of reduction: Addition of hydrogen or removal of oxygen • Addition of electrons 9:45 AM 2 Reducible Functional Groups 9:45 AM 3 Categories of Common Reducing Agents 9:45 AM 4 Relative Reactivity of Nucleophiles at the Reducible Functional Groups In the absence of any secondary interactions, the carbonyl compounds exhibit the following order of reactivity at the carbonyl This order may however be reversed in the presence of unique secondary interactions inherent in the molecule; interactions that may 9:45 AM be activated by some property of the reacting partner 5 Common Reducing Agents (Borohydrides) Reduction of Amides to Amines 9:45 AM 6 Common Reducing Agents (Borohydrides) Reduction of Carboxylic Acids to Primary Alcohols O 3 R CO2H + BH3 R O B + 3 H 3 2 Acyloxyborane 9:45 AM 7 Common Reducing Agents (Sodium Borohydride) The reductions with NaBH4 are commonly carried out in EtOH (Serving as a protic solvent) Note that nucleophilic attack occurs from the least hindered face of the 8 carbonyl Common Reducing Agents (Lithium Borohydride) The reductions with LiBH4 are commonly carried out in THF or ether Note that nucleophilic attack occurs from the least hindered face of the 9:45 AM 9 carbonyl. Common Reducing Agents (Borohydrides) The Influence of Metal Cations on Reactivity As a result of the differences in reactivity between sodium borohydride and lithium borohydride, chemoselectivity of reduction can be achieved by a judicious choice of reducing agent. 9:45 AM 10 Common Reducing Agents (Sodium Cyanoborohydride) 9:45 AM 11 Common Reducing Agents (Reductive Amination with Sodium Cyanoborohydride) 9:45 AM 12 Lithium Aluminium Hydride Lithium aluminiumhydride reacts the same way as lithium borohydride. -

UNIT 6 ELEMENTS of GROUP 13 Structure 6.1 Introduction Objectives I 6.2 Oailcrence, Extraction and Uses Occurrec - Extraction Uses I 6.3 General Characteristics
UNIT 6 ELEMENTS OF GROUP 13 structure 6.1 Introduction Objectives I 6.2 Oailcrence, Extraction and Uses Occurrec - Extraction uses i 6.3 General Characteristics I - 6.5 Halides of Bpron anel Aluminium Halides of Boron Halides of Aluminium 6.6 Oxides of Boron and Aluminium I Boric Oxide Aluminium Oxide 6.7 Oxoacids of Boron and Borates 6.8 Borazine 6.9 Complexation Behaviour 6.10 Anomalous Behaviour of Boron 6.11 Summary 6.12 Terminal Questions 6.13- Answers ' -- 6.1 INTRODUCTION In the previous two units, you studied the main features of the chemistry of Group 1 and Group 2 elements, i.e. the alkali and the alkaline earth metals. In this unit you - will study the elements of Group 13, namely, boron, aluminium, gallium, indium and, thallium. While studying the alkali and alkaline earth metals, you have seen that all Zhe elements of these two groups are highly reactive metals and the first element of each group shows some differences from the rest. In Group 13, the differences between the first element and the remaining elements become so pronounced that the first member of the group, i.e. boron is a nonmetal wheieas the rest of the elements are distinctly metallic in nature. In a way, this is the first group of the periodic table in which you observe a marked change in the hature of the elements . down the group. describe the chemistry of hydrides, halides and oxides of boron and aluminium, elucidate the structures of hydrides of boron and aluminium, 6.2 OCCURRENC~,EXTRACTION AND USES Elements bf Group 13 are sufficiently reactive. -

The Chemistry of Professor Craig Jon Hawker Born on January 11, 1964 in Queensland, Australia
The Chemistry of Professor Craig Jon Hawker Born on January 11, 1964 in Queensland, Australia Education University of Queensland, Australia. B.Sc. 1981-1984 Cambridge University, UK. Ph.D. 1985-1988 Professor Sir A.R. Battersby (Biosynthesis of Vitamin B12 – Model Studies) Cornell University, Postdoc 1988-1990 Professor J.M.J. Fréchet (dendrimer synthesis) Academic Appointments Queen Elizabeth II Research Fellow – University of Queensland. 1990-1993 Research Staff Member – IBM Almaden Research Center. 1993-2004 Professor of Materials, Chemistry, and Biochemistry – UCSB, 2004-2016 Clarke Professor – UCSB, 2013-Present Director, Dow Materials Institute – UCSB, 2013-Present Craig Jon Hawker Ph.D. Director, California Nanosystems Institute – UCSB, 2013-Present Research Interests: synthetic polymer chemistry, Mentored around 200 Graduate Students and Postdocs nanomaterials, dendrimer, Recipient of dozens of awards radical polymerization, Approximately 450 publications and 50 patents. biomaterials. Founder of Olaplex LLC and Tricida, Inc. The Chemistry of Professor Craig Jon Hawker - Outline I. Dendrimer Chemistry II. Nitroxide & Alkoxyamine Mediated “Living” Radical Polymerization III. “Living” Radical Polymerization by Light IV. Click Chemistry in Polymerizations and Bioapplications V. Industrial Success and Olaplex J. Polym. Sci. Part A: Polym. Chem. 2002, 40, 2719. Dendrimer Synthesis: Divergent Approach Reactive groups increases exponentially after each generation. Incomplete reaction at chain terminal lead to imperfections or failure sequences in the next generation. Large excess of reagents required in latter stages. Tomalia, D. A, et al. Polym. J. 1985, 17, 117. Mixture of inseparable multi-generation dendrimers. Tomalia, D. A, et al. Macromolecules 1986, 19, 2466. Dendrimer Synthesis: Convergent Approach Hawker, C. J.; Frechet, J. M. J. J. -

Prof. Colin Raston
Colin Raston, Ph.D ([email protected]) Professor Clean Technology, College of Science and Engineering Flinders University Adelaide, Australia Colin Raston is a Professor in Clean Technology, at Flinders University. He completed a PhD at the University of Western Australia, and after postdoctoral studies at the University of Sussex he was appointed a lecturer at The University of Western Australia (1981) then to positions as Professor of Chemistry at Griffith University (1988), Monash University (1995), The University of Leeds (2001), and The University of Western Australia (2003). He is a former President, Queensland Branch President, and Chair of the Inorganic Division, of the Royal Australian Chemical Institute (RACI). He has received the RACI’s Green Chemistry Challenge Award, the H.G. Smith Award for the most distinguished contribution to research in chemistry in Australia in the previous ten years, the Burrows Award for distinguished research in inorganic chemistry, and the Leighton Memorial Award for outstanding contributions to the profession. In 2015 he shared the Ig Nobel Prize in Chemistry with colleagues at the University of California, Irvine and the University of Western Australia, in 2016 he was Appointed an Officer in of the Order of Australia, and 2018 he was elected Fellow of the Australian Academy of Science. Prof Raston is former recipient of an Australian Research Council Special Investigator Award, two Senior Research Fellowships and two Australian Professorial Fellowships. His current research covers clean technology and green chemistry, nanotechnology and self-assembly, and is currently on the editorial advisory board of the international journals Green Chemistry, Crystal Growth and Design and Nature’s Scientific Reports. -

Novel Alkyne and Phosphaalkyne Coupling on an Ir4 Cluster: Synthesis and Molecular Structure of [Ir4(P-CO)(CO)7{P4-T13-Ph,PC(H)C(Ph)Pcbut}(P-Pph2)1 Maria Helena A
View Article Online / Journal Homepage / Table of Contents for this issue J. CHEM. SOC., CHEM. COMMUN., 1994 1869 Novel Alkyne and Phosphaalkyne Coupling on an Ir4 Cluster: Synthesis and Molecular Structure of [Ir4(p-CO)(CO)7{p4-t13-Ph,PC(H)C(Ph)PCBut}(p-PPh2)1 Maria Helena A. Benvenutti,asb Peter B. Hitchcock,b John F. Nixon*b and Maria D. Vargas*d a lnstituto de Quimica, Universidade Estadual de Campinas, CP 6154, Campinas, 13083, SP, Brazil b School of Chemistry and Moiecular Sciences, University of Sussex, Brighton, UK BN 1 9QJ The cluster compound [(p-H)Ir4(CO),(Ph2PCCPh)(p-PPh2)]1 reacts with the phosphaalkyne ButCP to yield [Ir4(p-CO)(CO),{p~-113-Ph2PC(H)C(Ph)PCBut}(p-PPh2)]3, containing the novel 2-phosphabutadienylphosphine fragment as a result of the coupling of ButCP with the diphenylphosphinoalkyne ligand and incorporation of the cluster bound H atom. There are relatively few examples of controlled alkyne-alkyne between the Ph2PCCPh ligand and the ButCP molecule, and coupling reactions at polynuclear carbonyl clusters.' The hydride migration to the resulting new phosphorus carbon chemistry of phosphaalkynes, RCP, is of considerable con- chain were established by 'H, 31P and 13C NMR spectroscopy. temporary interest and their similarity to alkynes has been In spite of the detailed spectroscopic studies undertaken, it stressed previously.2 There is only one reported interaction was impossible to establish unambiguously the position of the between an alkyne and a phosphaalkyne, leading to a hydrogen atom in the chain, and whether the diphenylphos- mononuclear 774-1-phosphacyclobutadienecomplex described phinoalkyne had undergone P-C,, bond cleavage. -

Intermediate Oxidation State Tungsten Acetylacetonate Complexes
Intermediate Oxidation State Tungsten Acetylacetonate Complexes Chetna Khosla A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Chemistry. Chapel Hill 2009 Approved by: Advisor: Professor Joseph L. Templeton Reader: Professor Maurice Brookhart Reader: Professor Cynthia Schauer Professor Valerie Ashby Professor H. Holden Thorp ABSTRACT Chetna Khosla: INTERMEDIATE OXIDATION STATE TUNGSTEN ACETYLACETONATE COMPLEXES (Under the direction of Professor Joseph L. Templeton) Previous work with tungsten(II) acetylacetonate complexes has focused on synthesis of complexes with -bound ligands (alkyne, nitrile, imine, ketone, aldehyde). In this work, 2 addition of methyl triflate (MeOTf) to W(CO)(acac)2(η -N≡CPh) produces the iminoacyl 2 complex [W(CO)acac)2(η -MeN=CPh)][OTf]. Displacement of the carbon monoxide in 2 + [W(CO)(acac)2(η -MeN≡CPh)] by isonitriles, bulky phosphines, and alkynes has been accomplished. The substitution of carbon monoxide by alkyne relieves the iminoacyl ligand of its role as a four-electron donor and enables further reduction of the C-N based ligand. Use 2 + of Na[HB(OMe)3] as a hydride source that attacks [W(RC≡CR′)(acac)2(η -MeN=CPh)] 2 yields the imine complex W(RC≡CR′)(acac)2(η -MeN=CHPh) with a diastereoselective ratio of 2:1. Addition of MeOTf leads to the final tungsten(II) iminium complex 2 [W(RC≡CR′)(acac)2(η -Me2N=CHPh)][OTf]. 2 Addition of m-chloroperoxybenzoic acid (MCPBA) to W(CO)(acac)2(η -N≡CR) 2 2 results in oxidation of the metal center to form the W(IV) d metal complex W(O)(acac)2(η - N≡CR). -

March 2010 Booklet
DPOLYMarch 2010 Meeting Program including Soft-Matter Physics sessions TM 2010 APS March Meeting • March 15–19 • Portland, Oregon DPOLY Short Course Polymers for Energy Generation and Storage March 13, 1:00 - 5:30 and March 14, 8:30 – 5:30 Oregon Convention Center Course Description Polymers hold much promise as active layers in inexpensive, lightweight energy generating and storing devices. Although the solid state physics and electrochemistry of such devices have been the subject of intense research, it has more recently become clear that an understanding of the polymer physics will lead to both an ability to control nanoscale morphology and optimize transport across the material. The purpose of this short course is to provide a background in the basic device physics of both organic photovoltaics and batteries to an audience primarily consisting of graduate students, postdoctoral researchers, and early career scientists already knowledgeable in polymer physics. It is hoped that this forum will both provide a basic foundation of knowledge as well as a deeper discussion of outstanding problems and avenues of research in energy relevant polymers. Each half of the course will begin by covering the basic underlying physics of charge/ion transport as well as device operation. Then our current understanding of the thermodynamics, morphology, self-assembly, and mechanisms of charge transport within these systems will be outlined with significant time reserved for discussion of gaps in current understanding and promising areas of future research. The course schedule allows for talks from each of the below speakers with ample time for discussion and interaction. This is roughly the schedule that has been followed by recent DPOLY short courses. -

Lowcoordinated Silicon and Hypercoordinated Carbon
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 557 Lowcoordinated Silicon and Hypercoordinated Carbon Structure and Stability of Silicon Analogs of Alkenes and Carbon Analogs of Silicates ANDERS M. EKLÖF ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6214 UPPSALA ISBN 978-91-554-7294-8 2008 urn:nbn:se:uu:diva-9298 !" # $ % #" # " & ' & & (' ') *' + , ') ,-.& / ) #) 0 + $ ) $ $ & $ / & /- / & $) / ) ""!) " ) ) 1$2 3!434""54!#354) 6 ' + 547 ' & ) *' ' & & $8498: ; ) ) *' & +' ' ' ' ' $<4 =*> & & ) ?<?* ' & ' ' & ) - & ; & ' ' ; & ' ' 4 4' ' 4 4 ; ' ' ' & + ' ; ' - ) @ ' & ' ' & % ' $ ') A ; #4 4 #4' 4 #4B24424'C4 & ' & ' ) 1 + ' + & ' ' ' & D4 E ; ' + & ' -4 ' ' && & ' ; ) 1 & ' E 4 & 4 & 4 4#4 +'' ' ' & D4 E ; ' ' ' & ; - ' ; 4 ) 1 & ' 5:5:25: 2 5: ' & $ 4 " 7 " 7 " $ #4$?4 $? #4 ) + ' ' 7 " 7 ' ' '4 + ' ) ;+ ; ' ' ' ' + !"#$ % & $ %$ % ' ()*$ $ !+)(,-. $ F / ) ,-.& # 1$$2 7"47#5 1$2 3!434""54!#354 43#3 B' << )-)< G 9 43#3C EXPERIMENTALISTS THINK SILICON IS REALLY FUN TO USE ITS PLACE IN NOVEL COMPOUNDS IS CERTAIN TO AMUSE THEY SIT ALL DAY IN LABORATORIES MAKING ALL THIS SLUDGE "LOADED WITH THE SILICON -

Fall 2017 PPG Fellow and of Mechanical Engineering
University of Michigan College of Engineering Rackham Graduate School Macro Messenger VOLUME XXVIII 2017 The Newsletter of the Director Mark Banaszak Holl’s Message Macromolecular Science and Engineering Program Dear Alumni and Friends, I hope you had as exciting, fun, and productive a year as our Macro graduate students! I am delighted to share some highlights from the past few months. In November I traveled around the world with four Macro students for a scientific and cultural exchange at the Nara Institute of Science & Technology (NAIST). Two NAIST faculty and six students had visited for the Macro symposium in October and we were excited to have the opportunity to visit their institution. Modern day science and engineering challenges are global in nature and impact, and we are delighted to be continuing these important efforts initiated by former Director Rick Laine. Our interactions and collaborations with industry continued to grow this term. The Director’s Message..1-2 ACS POLY/PMSE student chapter hosted campus visits by number companies as well Macro News...........3 as site visits to local company labs. We were also delighted to receive summer and Symposium.........4-7 fall fellowship support from 3M and PPG for our Ph.D. program. Their contributions Research News......8-11 provided support to seven Macro students. These interactions and support are Faculty News........12 immensely important for training the next generation of scientists and engineers. Student News......12-15 Giving................16 Our 41st Annual Symposium, “Emergent Polymer Science & Engineering” was organized by Professors Kenichi Kuroda and Tim Scott in addition to Ph.D.