Negative Breast Cancers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Downloaded the “Top Edge” Version
bioRxiv preprint doi: https://doi.org/10.1101/855338; this version posted December 6, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Drosophila models of pathogenic copy-number variant genes show global and 2 non-neuronal defects during development 3 Short title: Non-neuronal defects of fly homologs of CNV genes 4 Tanzeen Yusuff1,4, Matthew Jensen1,4, Sneha Yennawar1,4, Lucilla Pizzo1, Siddharth 5 Karthikeyan1, Dagny J. Gould1, Avik Sarker1, Yurika Matsui1,2, Janani Iyer1, Zhi-Chun Lai1,2, 6 and Santhosh Girirajan1,3* 7 8 1. Department of Biochemistry and Molecular Biology, Pennsylvania State University, 9 University Park, PA 16802 10 2. Department of Biology, Pennsylvania State University, University Park, PA 16802 11 3. Department of Anthropology, Pennsylvania State University, University Park, PA 16802 12 4 contributed equally to work 13 14 *Correspondence: 15 Santhosh Girirajan, MBBS, PhD 16 205A Life Sciences Building 17 Pennsylvania State University 18 University Park, PA 16802 19 E-mail: [email protected] 20 Phone: 814-865-0674 21 1 bioRxiv preprint doi: https://doi.org/10.1101/855338; this version posted December 6, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 22 ABSTRACT 23 While rare pathogenic copy-number variants (CNVs) are associated with both neuronal and non- 24 neuronal phenotypes, functional studies evaluating these regions have focused on the molecular 25 basis of neuronal defects. -

Overexpressed HSF1 Cancer Signature Genes Cluster in Human Chromosome 8Q Christopher Q
Zhang et al. Human Genomics (2017) 11:35 DOI 10.1186/s40246-017-0131-5 PRIMARY RESEARCH Open Access Overexpressed HSF1 cancer signature genes cluster in human chromosome 8q Christopher Q. Zhang1,4, Heinric Williams3,4, Thomas L. Prince3,4† and Eric S. Ho1,2*† Abstract Background: HSF1 (heat shock factor 1) is a transcription factor that is found to facilitate malignant cancer development and proliferation. In cancer cells, HSF1 mediates a set of genes distinct from heat shock that contributes to malignancy. This set of genes is known as the HSF1 Cancer Signature genes or simply HSF1-CanSig genes. HSF1-CanSig genes function and operate differently than typical cancer-causing genes, yet it is involved in fundamental oncogenic processes. Results: By utilizing expression data from 9241 cancer patients, we identified that human chromosome 8q21-24 is a location hotspot for the most frequently overexpressed HSF1-CanSig genes. Intriguingly, the strength of the HSF1 cancer program correlates with the number of overexpressed HSF1-CanSig genes in 8q, illuminating the essential role of HSF1 in mediating gene expression in different cancers. Chromosome 8q21-24 is found under selective pressure in preserving gene order as it exhibits strong synteny among human, mouse, rat, and bovine, although the biological significance remains unknown. Statistical modeling, hierarchical clustering, and gene ontology-based pathway analyses indicate crosstalk between HSF1-mediated responses and pre-mRNA 3′ processing in cancers. Conclusions: Our results confirm the unique role of chromosome 8q mediated by the master regulator HSF1 in cancer cases. Additionally, this study highlights the connection between cellular processes triggered by HSF1 and pre-mRNA 3′ processing in cancers. -

393LN V 393P 344SQ V 393P Probe Set Entrez Gene
393LN v 393P 344SQ v 393P Entrez fold fold probe set Gene Gene Symbol Gene cluster Gene Title p-value change p-value change chemokine (C-C motif) ligand 21b /// chemokine (C-C motif) ligand 21a /// chemokine (C-C motif) ligand 21c 1419426_s_at 18829 /// Ccl21b /// Ccl2 1 - up 393 LN only (leucine) 0.0047 9.199837 0.45212 6.847887 nuclear factor of activated T-cells, cytoplasmic, calcineurin- 1447085_s_at 18018 Nfatc1 1 - up 393 LN only dependent 1 0.009048 12.065 0.13718 4.81 RIKEN cDNA 1453647_at 78668 9530059J11Rik1 - up 393 LN only 9530059J11 gene 0.002208 5.482897 0.27642 3.45171 transient receptor potential cation channel, subfamily 1457164_at 277328 Trpa1 1 - up 393 LN only A, member 1 0.000111 9.180344 0.01771 3.048114 regulating synaptic membrane 1422809_at 116838 Rims2 1 - up 393 LN only exocytosis 2 0.001891 8.560424 0.13159 2.980501 glial cell line derived neurotrophic factor family receptor alpha 1433716_x_at 14586 Gfra2 1 - up 393 LN only 2 0.006868 30.88736 0.01066 2.811211 1446936_at --- --- 1 - up 393 LN only --- 0.007695 6.373955 0.11733 2.480287 zinc finger protein 1438742_at 320683 Zfp629 1 - up 393 LN only 629 0.002644 5.231855 0.38124 2.377016 phospholipase A2, 1426019_at 18786 Plaa 1 - up 393 LN only activating protein 0.008657 6.2364 0.12336 2.262117 1445314_at 14009 Etv1 1 - up 393 LN only ets variant gene 1 0.007224 3.643646 0.36434 2.01989 ciliary rootlet coiled- 1427338_at 230872 Crocc 1 - up 393 LN only coil, rootletin 0.002482 7.783242 0.49977 1.794171 expressed sequence 1436585_at 99463 BB182297 1 - up 393 -

The C. Elegans 3′ Utrome V2 Resource for Studying Mrna Cleavage and Polyadenylation, 3′-UTR Biology, and Mirna Targeting
Downloaded from genome.cshlp.org on October 8, 2021 - Published by Cold Spring Harbor Laboratory Press Resource The C. elegans 3′ UTRome v2 resource for studying mRNA cleavage and polyadenylation, 3′-UTR biology, and miRNA targeting Hannah S. Steber,1,2 Christina Gallante,3 Shannon O’Brien,2 Po-Lin Chiu,4 and Marco Mangone1,2,5 1Molecular and Cellular Biology Graduate Program, School of Life Sciences, Tempe, Arizona 85287; 2Virginia G. Piper Center for Personalized Diagnostics, The Biodesign Institute at Arizona State University, Tempe, Arizona 85281, USA; 3Barrett, The Honors College, Arizona State University, Tempe, Arizona 85281, USA; 4Center for Applied Structural Discovery, The Biodesign Institute at Arizona State University, Tempe, Arizona 85287, USA 3′ Untranslated regions (3′ UTRs) of mRNAs emerged as central regulators of cellular function because they contain im- portant but poorly characterized cis-regulatory elements targeted by a multitude of regulatory factors. The model nema- tode Caenorhabditis elegans is ideal to study these interactions because it possesses a well-defined 3′ UTRome. To improve its annotation, we have used a genome-wide bioinformatics approach to download raw transcriptome data for 1088 transcrip- tome data sets corresponding to the entire collection of C. elegans trancriptomes from 2015 to 2018 from the Sequence Read Archive at the NCBI. We then extracted and mapped high-quality 3′-UTR data at ultradeep coverage. Here, we describe and release to the community the updated version of the worm 3′ UTRome, which we named 3′ UTRome v2. This resource contains high-quality 3′-UTR data mapped at single-base ultraresolution for 23,084 3′-UTR isoform variants corresponding to 14,788 protein-coding genes and is updated to the latest release of WormBase. -

Selectivity of ORC Binding Sites and the Relation to Replication Timing, Fragile Sites, and Deletions in Cancers
Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers Benoit Miottoa,b,c,d,1, Zhe Jia,e,1, and Kevin Struhla,2 aDepartment of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA 02115; bINSERM, U1016, Institut Cochin, 75014 Paris, France; cCNRS, UMR8104, 75014 Paris, France; dUniversite Paris Descartes, Sorbonne Paris Cite, 75006 Paris, France; and eBroad Institute of MIT and Harvard, Cambridge, MA 02142 Contributed by Kevin Struhl, June 14, 2016 (sent for review April 27, 2016; reviewed by Bing Ren and Nicholas Rhind) The origin recognition complex (ORC) binds sites from which DNA regions are replicated late (18–20). One possible explanation for the replication is initiated. We address ORC binding selectivity in vivo by replication timing pattern is that origins are programmed to generate mapping ∼52,000 ORC2 binding sites throughout the human genome. the same pattern in all cells. Alternatively, origins could fire sto- The ORC binding profile is broader than those of sequence-specific chastically so that the pattern varies from cell to cell. Early versions transcription factors, suggesting that ORC is not bound or recruited of the stochastic firing model invoked functional differences between to specific DNA sequences. Instead, ORC binds nonspecifically to open early versus late origins. However, the finding of more ORC binding (DNase I-hypersensitive) regions containing active chromatin marks sites in early replicating regions of the Drosophila genome suggested such as H3 acetylation and H3K4 methylation. ORC sites in early and the possibility that the timing pattern might arise from stochastic late replicating regions have similar properties, but there are far more firing from all origins (4, 5). -

A Plant-Like Mechanism Coupling M6a Reading to Polyadenylation
RESEARCH ARTICLE A plant-like mechanism coupling m6A reading to polyadenylation safeguards transcriptome integrity and developmental gene partitioning in Toxoplasma Dayana C Farhat1, Matthew W Bowler2, Guillaume Communie3, Dominique Pontier4, Lucid Belmudes5, Caroline Mas6, Charlotte Corrao1, Yohann Coute´ 5, Alexandre Bougdour1, Thierry Lagrange4, Mohamed-Ali Hakimi1†*, Christopher Swale1†* 1IAB,Team Host-Pathogen Interactions & Immunity to Infection, INSERMU1209, CNRSUMR5309, Grenoble Alpes University, Grenoble, France; 2European Molecular Biology Laboratory, Grenoble, France; 3Institut Laue-Langevin, Grenoble, France; 4Laboratoire Genome et Developpement des Plantes (LGDP), UMR5096, Centre National de la Recherche Scientifique (CNRS), Universitede Perpignan via Domitia (UPVD), Perpignan, France; 5Univ. Grenoble Alpes, INSERM, CEA, UMR BioSante´ U1292, CNRS, CEA, Grenoble, France; 6Integrated Structural Biology Grenoble (ISBG) CNRS, CEA, Universite´ Grenoble Alpes, EMBL, Grenoble, France *For correspondence: [email protected] Abstract Correct 3’end processing of mRNAs is one of the regulatory cornerstones of gene (M-AH); expression. In a parasite that must adapt to the regulatory requirements of its multi-host life style, christopher.swale@univ-grenoble- there is a need to adopt additional means to partition the distinct transcriptional signatures of the alpes.fr (CS) closely and tandemly arranged stage-specific genes. In this study, we report our findings in T. †These authors contributed gondii of an m6A-dependent 3’end polyadenylation serving as a transcriptional barrier at these loci. equally to this work We identify the core polyadenylation complex within T. gondii and establish CPSF4 as a reader for Competing interests: The m6A-modified mRNAs, via a YTH domain within its C-terminus, a feature which is shared with authors declare that no plants. -

Genome-Wide Association for Milk Production and Female Fertility Traits in Canadian Dairy Holstein Cattle Shadi Nayeri1, Mehdi Sargolzaei2,3, Mohammed K
Nayeri et al. BMC Genetics (2016) 17:75 DOI 10.1186/s12863-016-0386-1 RESEARCH ARTICLE Open Access Genome-wide association for milk production and female fertility traits in Canadian dairy Holstein cattle Shadi Nayeri1, Mehdi Sargolzaei2,3, Mohammed K. Abo-Ismail2,4, Natalie May1, Stephen P. Miller2,5, Flavio Schenkel2, Stephen S. Moore1,6 and Paul Stothard1* Abstract Background: Genome-wide association studies (GWAS) are a powerful tool for detecting genomic regions explaining variation in phenotype. The objectives of the present study were to identify or refine the positions of genomic regions affecting milk production, milk components and fertility traits in Canadian Holstein cattle, and to use these positions to identify genes and pathways that may influence these traits. Result: Several QTL regions were detected for milk production (MILK), fat production (FAT), protein production (PROT) and fat and protein deviation (FATD, PROTD respectively). The identified QTL regions for production traits (including milk production) support previous findings and some overlap with genes with known relevant biological functions identified in earlier studies such as DGAT1 and CPSF1. A significant region on chromosome 21 overlapping with the gene FAM181A and not previous linked to fertility in dairy cattle was identified for the calving to first service interval and days open. A functional enrichment analysis of the GWAS results yielded GO terms consistent with the specific phenotypes tested, for example GO terms GO:0007595 (lactation) and GO:0043627 (response to estrogen) for milk production (MILK), GO:0051057 (positive regulation of small GTPase mediated signal transduction) for fat production (FAT), GO:0040019 (positive regulation of embryonic development) for first service to calving interval (CTFS) and GO:0043268 (positive regulation of potassium ion transport) for days open (DO). -
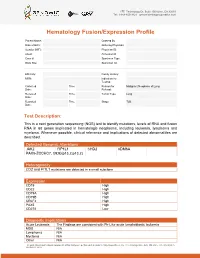
Sample Report for Hematology Fusion/Expression Profile
17527 Technology Dr, Suite 100 Irvine, CA 92618 Tel: 1-949-450-9421 genomictestingcooperative.com Hematology Fusion/Expression Profile Patient Name: Ordered By Date of Birth: Ordering Physician: Gender (M/F): Physician ID: Client: Accession #: Case #: Specimen Type: Body Site: Specimen ID: _____________________________________________________________________________________________ Ethnicity: Family History: MRN: Indication for Testing: Collected Time Reason for Malignant Neoplasm of Lung Date: : Referral: Received Time Tumor Type: Lung Date: : Reported Time Stage: T2B Date: : Test Description: This is a next generation sequencing (NGS) test to identify mutations, levels of RNA and fusion RNA in 68 genes implicated in hematologic neoplasms, including leukemia, lymphoma and myeloma. Whenever possible, clinical relevance and implications of detected abnormalities are described. Detected Genomic Alterations JAK2 FIP1L1 CHD2 KDM6A PAX5-ZCCHC7, t(9;9)(p13.2;p13.2) Heterogeneity CD2 and FI1L1 mutations are detected in a small subclone Expression CD19 High CD22 High CD79A High CD79B High CRLF2 High PAX5 High CD274 Low Diagnostic Implications Acute Leukemia The Findings are consistent with Ph-Like acute lymphoblastic leukemia MDS N/A Lymphoma N/A Myeloma N/A Other N/A The professional and technical components of this assay were performed at Genomic Testing Cooperative, LCA, 27 Technology Drive, Suite 100, Irvine, CA 92618 (CLIA ID: 05D2111917). The assay is FDA cleared and the performance characteristics were established at this location and was .... 27 Technology Dr, Suite 100 Irvine, CA 92618 Tel: 1-949-450-9421 genomictestingcooperative.com Therapeutic Implications JAK2 Momelotinib and ruxolitinib Prognostic Implications JAK2 Poor CRLF2 Poor Overall Poor Relevant Genes with No Alteration None Results Summary • Fusion (PAX5-ZCCHC7) transcript involving PAX5 and ZCCHC7 genes resulting from t(9;9)(p13.2;p13.2) translocation is detected. -

Dynamic Analyses of Alternative Polyadenylation from RNA-Seq Reveal a 30-UTR Landscape Across Seven Tumour Types
ARTICLE Received 11 Jul 2014 | Accepted 15 Sep 2014 | Published 20 Nov 2014 DOI: 10.1038/ncomms6274 Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 30-UTR landscape across seven tumour types Zheng Xia1,2, Lawrence A. Donehower3,4, Thomas A. Cooper2,5,6, Joel R. Neilson6, David A. Wheeler4,7, Eric J. Wagner8 & Wei Li1,2 Alternative polyadenylation (APA) is a pervasive mechanism in the regulation of most human genes, and its implication in diseases including cancer is only beginning to be appreciated. Since conventional APA profiling has not been widely adopted, global cancer APA studies are very limited. Here we develop a novel bioinformatics algorithm (DaPars) for the de novo identification of dynamic APAs from standard RNA-seq. When applied to 358 TCGA Pan- Cancer tumour/normal pairs across seven tumour types, DaPars reveals 1,346 genes with recurrent and tumour-specific APAs. Most APA genes (91%) have shorter 30-untranslated regions (30 UTRs) in tumours that can avoid microRNA-mediated repression, including glutaminase (GLS), a key metabolic enzyme for tumour proliferation. Interestingly, selected APA events add strong prognostic power beyond common clinical and molecular variables, suggesting their potential as novel prognostic biomarkers. Finally, our results implicate CstF64, an essential polyadenylation factor, as a master regulator of 30-UTR shortening across multiple tumour types. 1 Division of Biostatistics, Dan L Duncan Cancer Center, Baylor College of Medicine, Houston, Texas 77030, USA. 2 Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, Texas 77030, USA. 3 Department of Molecular Virology and Microbiology, Baylor College of Medicine, Houston, Texas 77030, USA. -

Identification of High-Confidence RNA Regulatory Elements By
Li et al. Genome Biology (2017) 18:169 DOI 10.1186/s13059-017-1298-8 METHOD Open Access Identification of high-confidence RNA regulatory elements by combinatorial classification of RNA–protein binding sites Yang Eric Li1†, Mu Xiao2†, Binbin Shi1†, Yu-Cheng T. Yang1, Dong Wang1, Fei Wang2, Marco Marcia3 and Zhi John Lu1* Abstract Crosslinking immunoprecipitation sequencing (CLIP-seq) technologies have enabled researchers to characterize transcriptome-wide binding sites of RNA-binding protein (RBP) with high resolution. We apply a soft-clustering method, RBPgroup, to various CLIP-seq datasets to group together RBPs that specifically bind the same RNA sites. Such combinatorial clustering of RBPs helps interpret CLIP-seq data and suggests functional RNA regulatory elements. Furthermore, we validate two RBP–RBP interactions in cell lines. Our approach links proteins and RNA motifs known to possess similar biochemical and cellular properties and can, when used in conjunction with additional experimental data, identify high-confidence RBP groups and their associated RNA regulatory elements. Keywords: RNA-binding protein, CLIP-seq, Non-negative matrix factorization, RBPgroup Background protein binding sites with high resolution in different RNA-binding proteins (RBPs) are essential to sustain mammalian cells [12–14]. The CLIP-seq data of mul- fundamental cellular functions, such as splicing, polya- tiple RNA binding proteins have been curated and denylation, transport, translation, and degradation of annotated in specific databases, such as CLIPdb, RNA transcripts [1, 2]. One study estimated that more POSTAR, and STARbase [15–17]. Several significant than 1500 different RBPs exist in human [3]. These studies improved the prediction of individual RBPs’ RBPs cooperate or compete with each other in binding binding sites by training on CLIP-seq and RNAcompete their RNA targets [4–6]. -
Supporting Information
Supporting Information: “Proxy-Phenotype Method Identifies Common Genetic Variants Associated with Cognitive Performance” __________________________________________________________________________________________ This document provides further details about materials, methods and additional analyses to accompany the research report “Proxy-Phenotype Method Identifies Common Genetic Variants Associated with Cognitive Performance.” 1 Contents Materials and Methods ......................................................................................................................... 3 1. META-ANALYSES AND SELECTION OF EDUCATION-ASSOCIATED CANDIDATE SNPS .................................. 3 2. COGNITIVE PERFORMANCE SAMPLE ......................................................................................................... 3 3. COGNITIVE PERFORMANCE MEASURES ...................................................................................................... 4 4. GENOTYPING AND IMPUTATION ................................................................................................................ 6 5. QUALITY CONTROL ................................................................................................................................... 7 6. ASSOCIATION ANALYSIS ........................................................................................................................... 7 7. META-ANALYSIS ...................................................................................................................................... -

Gene Section Review
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL INIST-CNRS Gene Section Review FIP1L1 (factor interacting with PAPOLA and CPSF1) Adriana Zamecnikova, Soad Al Bahar Kuwait Cancer Control Center, Department of Hematology, Laboratory of Cancer Genetics, Kuwait Published in Atlas Database: November 2014 Online updated version : http://AtlasGeneticsOncology.org/Genes/FIP1L1ID40577ch4q12.html Printable original version : http://documents.irevues.inist.fr/bitstream/handle/2042/62487/11-2014-FIP1L1ID40577ch4q12.pdf DOI: 10.4267/2042/62487 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2015 Atlas of Genetics and Cytogenetics in Oncology and Haematology binding motif (Preker et al., 1995; Kaufmann et al., Abstract 2004). Review on FIP1L1, with data on DNA/RNA, on the Size: 4 isoforms as a result of splicing processes; the protein encoded and where the gene is implicated. larger transcript has 594 amino acids (molecular Keywords weight 66526 Da). In the second isoform (559 amino acids; 63048 Da), exon 9 is deleted. The third FIP1L1; factor interacting with PAPOLA and isoform is the shortest (378 amino acids; molecular CPSF1; cleavage and polyadenylation specificity weight 40835 Da) due to the deletion of some exons factor complex; RNA-binding. (exon 2 and 14-18) during splicing processes. In the Identity last isoform, exons 2, 9 and 11 are deleted. Expression Other names: FIP1, Rhe, hFip1 FIP1L1 expression is present in different immunitary HGNC (Hugo): FIP1L1 cells, embrionic and fetal cells, reproductive tissues, Location: 4q12 in blood and bone marrow and others. Note Localisation Recommended name: Pre-mRNA 3'-end-processing Subcellular location: Nucleus.