Useful Reactions from CHM1C3 and CHM2C3-A of BIRMINGHAM
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Nucleophilic Aromatic Substitution
NUCLEOPHILIC AROMATIC SUBSTITUTION Ms. Prerana Sanas M.Pharm (Pharmceutical Chemistry) Asst.Professor, NCRD’s Sterling Institute of Pharmacy Nerul Navi Mumbai Nucleophilic aromatic substitution results in the substitution of a halogen X on a benzene ring by a nucleophile (:Nu– ). Aryl halides undergo a limited number of substitution reactions with strong nucleophiles. NAS occurs by two mechanisms i) Bimoleccular displacement (Addition –Elimination) ii) Benzyne Formation( Elimination –Addition) 7/5/2019 Ms.Prerana Sanas 2 Bimolecular displacement (Addition – Elimination) Aryl halides with strong electron-withdrawing groups (such as NO2) on the ortho or para positions react with nucleophiles to afford substitution products. For example, treatment of p-chloronitrobenzene with hydroxide (– OH) affords p-nitrophenol by replacement of Cl by OH. Nucleophilic aromatic substitution occurs with a variety of strong nucleophiles, including – OH, – OR, – NH2, – SR, and in some cases, neutral nucleophiles such as NH3 and RNH2 . 7/5/2019 Ms.Prerana Sanas 3 Mechanism…… The mechanism of these reactions has two steps: Step i) Addition of the nucleophile (:Nu– ) forms a resonance-stabilized carbanion with a new C – Nu bond—three resonance structures can be drawn. • Step [1] is rate-determining since the aromaticity of the benzene ring is lost. In Step ii) loss of the leaving group re-forms the aromatic ring. This step is fast because the aromaticity of the benzene ring is restored. 7/5/2019 Ms.Prerana Sanas 4 Factors affecting Bimolecular displacement Increasing the number of electron-withdrawing groups increases the reactivity of the aryl halide. Electron-withdrawing groups stabilize the intermediate carbanion, and by the Hammond postulate, lower the energy of the transition state that forms it. -

Lecture 5 Diastereoselective Addition Into Carbonyl Compounds
Lecture 5 Diastereoselective Addition into Carbonyl Compounds Containing α-Stereogenic Centres Learning outcomes: by the end of this lecture, and after answering the associated problems, you will be able to: 1. use the Felkin-Anh T.S. to predict the stereochemical outcome of reactions carried out on carbonyl compounds that possess an α-stereogenic centre; 2. rationalise the preferential adoption of a Felkin-Anh T.S. in nucleophilic addition reactions on steric and stereoelectronic grounds; 3. understand how the presence of an α-electron-withdrawing substituent affects the Felkin-Anh T.S.; 4. use the Felkin-Anh T.S. to prepare 1,2-syn diols from α-alkoxy ketones; 5. use the Cram chelation model to prepare 1,2-anti diols from α-alkoxy ketones. Stereoselective Addition of Nucleophiles into Ketones and Aldehydes containing α- Stereogenic Centres The addition of a nucleophile into a chiral ketone or aldehyde provides diastereoisomers. When the stereogenic centres in the substrate are close to the reacting carbonyl group (e.g. 1,2-disposed), then it is often possible to exploit this stereochemical information to control the stereoselectivity of the addition reaction. This method for controlling the stereochemical outcome of a reaction is known as substrate control. A number of models have been developed for predicting the stereochemical outcome of this type of reaction. Felkin-Anh Model Consider a carbonyl compound containing an α-stereogenic centre in which the three substituents at the α-site are well differentiated in size: O HO Nu Nu OH RL Nu RL RL R R R S M S M S RM R R R R R RS = small substituent RM = medium-sized substituent RL = large substituent Of the two diastereoisomeric alcohol addition products, one will be formed to a greater extent than the other. -

Ketenes 25/01/2014 Part 1
Baran Group Meeting Hai Dao Ketenes 25/01/2014 Part 1. Introduction Ph Ph n H Pr3N C A brief history Cl C Ph + nPr NHCl Ph O 3 1828: Synthesis of urea = the starting point of modern organic chemistry. O 1901: Wedekind's proposal for the formation of ketene equivalent (confirmed by Staudinger 1911) Wedekind's proposal (1901) 1902: Wolff rearrangement, Wolff, L. Liebigs Ann. Chem. 1902, 325, 129. 2 Wolff adopt a ketene structure in 1912. R 2 hν R R2 1905: First synthesis and characterization of a ketene: in an efford to synthesize radical 2, 1 ROH R C Staudinger has synthesized diphenylketene 3, Staudinger, H. et al., Chem. Ber. 1905, 1735. N2 1 RO CH or Δ C R C R1 1907-8: synthesis and dicussion about structure of the parent ketene, Wilsmore, O O J. Am. Chem. Soc. 1907, 1938; Wilsmore and Stewart Chem. Ber. 1908, 1025; Staudinger and Wolff rearrangement (1902) O Klever Chem. Ber. 1908, 1516. Ph Ph Cl Zn Ph O hot Pt wire Zn Br Cl Cl CH CH2 Ph C C vs. C Br C Ph Ph HO O O O O O O O 1 3 (isolated) 2 Wilsmore's synthesis and proposal (1907-8) Staudinger's synthesis and proposal (1908) wanted to make Staudinger's discovery (1905) Latest books: ketene (Tidwell, 1995), ketene II (Tidwell, 2006), Science of Synthesis, Vol. 23 (2006); Latest review: new direactions in ketene chemistry: the land of opportunity (Tidwell et al., Eur. J. Org. Chem. 2012, 1081). Search for ketenes, Google gave 406,000 (vs. -

Aromatic Nucleophilic Substitution Reaction
Aromatic Nucleophilic Substitution Reaction DR. RAJENDRA R TAYADE ASSISTANT PROFESSOR DEPARTMENT OF CHEMISTRY INSTITUTE OF SCIENCE, NAGPUR Principles There are four principal mechanisms for aromatic nucleophilic substitution which are similar to that of aliphatic nucleophilic substitution. (SN1, SN2, SNi, SET Mechanism) 1. SNAr Mechanism- addition / elimination CF3, CN, CHO, COR, COOH, Br, Cl, I Common Activating Groups for NAS Step [1] Addition of the nucleophile (:Nu–) to form a carbanion Addition of the nucleophile (:Nu–) forms a resonance-stabilized carbanion with a new C – Nu bond— three resonance structures can be drawn. • Step is rate-determining • Aromaticity of the benzene ring is lost Step [2] loss of the leaving group re-forms the aromatic ring. • This step is fast because the aromaticity of the benzene ring is restored. ? Explain why a methoxy group (CH3O) increases the rate of electrophilic aromatic substitution, but decreases the rate of nucleophilic aromatic substitution. 2.ArSN1 Mechanism- elimination /addition • This mechanism operates in the reaction of diazonium salts with nucleophiles. •The driving force resides in the strength of the bonding in the nitrogen molecule that makes it a particularly good leaving group. 3.Benzyne Mechanism- elimination /addition Step [1] Elimination of HX to form benzyne Elimination of H and X from two adjacent carbons forms a reactive benzyne intermediate Step [2] Nucleophilic addition to form the substitution product Addition of the nucleophile (–OH in this case) and protonation form the substitution product Evidence for the Benzyne Mechanism Trapping in Diels/Alder Reaction O O B E N Z Y N E C C O O O D i e l s / A l d e r O N H 3 N N Dienophile Diene A d d u c t Substrate Modification – absence of a hydrogens LG Substituent Substituent No Reaction Base Isotopic Labeling LG Nu H Nu Structure of Benzyne • The σ bond is formed by overlap of two sp2 hybrid orbitals. -
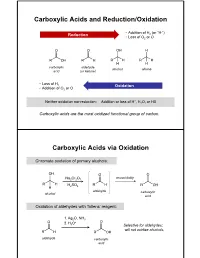
Carboxylic Acids and Reduction/Oxidation Carboxylic
Carboxylic Acids and Reduction/Oxidation • Addition of H (or “H-”) Reduction 2 • Loss of O2 or O carboxylic aldehyde alcohol alkane acid (or ketone) • Loss of H 2 Oxidation • Addition of O2 or O + Neither oxidation nor reduction: Addition or loss of H , H2O, or HX Carboxylic acids are the most oxidized functional group of carbon. Carboxylic Acids via Oxidation Chromate oxidation of primary alcohols: unavoidably Na2Cr2O7 H2SO4 aldehyde carboxylic alcohol acid Oxidation of aldehydes with Tollens’ reagent: 1. Ag2O, NH3 2. H O+ 3 Selective for aldehydes; will not oxidize alcohols. aldehyde carboxylic acid Carboxylic Acids via Oxidation Oxidative cleavage of alkynes: 1. O3 2. H2O internal alkyne Oxidation of benzylic carbons: KMnO4 Purifying Carboxylic Acids by Acid-Base Extraction Acidic and basic organic molecules can be separated from other substances by manipulating their protonation state and solubility. Let’s say we run a reaction. Na2Cr2O7 + unreacted H2SO4 + Na+ 3+ We want this… + Cr(H2O)6 + organic + HSO - …but not these. side products 4 How do we isolate our desired product from the other materials, without using distillation or chromatography? Acid-Base Extraction Step 1: Remove inorganics from organics via differential solubility. a separatory (sept) funnel - Na+ HSO4 Cr(H O) 3+ H2O 2 6 CHCl3 + organic side densities: products (H2O) = 1 g/mL (CHCl3) = 1.48 g/mL A large density difference ensures the two liquids separate completely and quickly. (Much faster than, say, oil and water.) Step 2: Add basic water to deprotonate carboxylic acid, and transfer it to the aqueous phase. - Na+ HSO4 3+ discard Cr(H2O)6 H2O layer + organic + organic side side products products add discard NaOH/ H2O CHCl3 layer + organic side products Step 3: Re-acidify carboxylate to return it to organic solution. -

Worksheet – Nucleophilic Addition and Substitution a Variety of Reagents Can Add Across the C=C of Alkenes, H-X, H-OH, Br-Br and H-H
Worksheet – Nucleophilic Addition and Substitution A variety of reagents can add across the C=C of alkenes, H-X, H-OH, Br-Br and H-H. When the non-symmetric species (H-X and H-OH) add, Markovnikov’s rule can be used to predict the major and minor products, based on the stability of the intermediate carbocations. Reagents, like H-OH and H-OR(alcohols) can add across the C=O bond of aldehydes and ketones. The acid catalyzed mechanism is very similar to the addition to alkenes, but in these reactions, the H always adds to the O and the nucleophile (:OH or :OR) adds to the carbon. 1. Write the mechanism for the hydration of 2-methylpropene: The electrons in the C=O double bond can be used in the same way. Because of the polarity of the C=O bonds, the H+ will add to the O and the O- H or O-R will add to the carbon. 2. Write the mechanism for the addition of water to 2-propanone: This product is called a “diol” or “hydrate” and is unstable. The same reaction can take place between an aldehyde and water. 3. Write the mechanism for the addition of ethanol to propanone: This product, also unstable, is called a hemi-ketal (ketone + alcohol). The same reaction takes place between an aldehyde and an alcohol forming a hemi-acetal. There are no more electrons, so no further addition products are possible. However, a second equivalent of alcohol can react with the hemi-acetals and hemi-ketals in a nucleophilic substitution reaction. -

Nucleophilic Addition to the Carbonyl Group 6
Nucleophilic addition to the carbonyl group 6 Connections Building on Arriving at Looking forward to • Functional groups, especially the C=O • How and why the C=O group reacts • Additions of organometallic group ch2 with nucleophiles reagents ch9 • Identifying the functional groups in a • Explaining the reactivity of the C=O • Substitution reactions of the C=O molecule spectroscopically ch3 group using molecular orbitals and curly group’s oxygen atom ch11 • How molecular orbitals explain arrows • How the C=O group in derivatives of molecular shapes and functional • What sorts of molecules can be made by carboxylic acids promotes substitution groups ch4 reactions of C=O groups reactions ch10 • How, and why, molecules react together • How acid or base catalysts improve the • C=O groups with an adjacent double and using curly arrows to describe reactivity of the C=O group bond ch22 reactions ch5 Molecular orbitals explain the reactivity of the carbonyl group We are now going to leave to one side most of the reactions you met in the last chapter—we will come back to them all again later in the book. In this chapter we are going to concentrate on just one of them—probably the simplest of all organic reactions—the addition of a nucleo- phile to a carbonyl group. The carbonyl group, as found in aldehydes, ketones, and many other compounds, is without doubt the most important functional group in organic chemis- try, and that is another reason why we have chosen it as our fi rst topic for more detailed study. You met nucleophilic addition to a carbonyl group on pp. -

Reaction-Mechanisms-GOC-Book.Pdf
158 Essential Notes on General Organic Chemistry (G.O.C.) CHAPTER Reaction Mechanisms 4 Topics Coverage :- • Substitution Reactions • Free Radical Reactions • SN1 Mechanism • SN2 Mechanism • SN1 v/s SN2 Mechanisms • E1 Mechanism • E2 Mechanism • E1 v/s E2 Mechanisms • Substitution v/s Elimination Reactions • Aromatic Substitution Electrophilic Mechanism • Aromatic Substitution Nucleophilic Mechanism • Addition Nucleophilic Mechanism • Addition Electrophilic Mechanism • Addition Free Radical Mechanism • RARE MECHANISMS : (SNi, E1cb, SN', NGP, Benzyne, Ipso) • Rearrangements Introduction : How exactly does an organic reaction take place is the subject of study in this chapter of Reaction Mechanisms. Just as detectives investigate a crime after it has taken place and arrive at a reasonable prediction about how the crime may have occurred, similarly organic chemists predict by a number of laboratory techniques using sophisticated instrumentation how an organic reaction may have taken place. Many of these techniques are based on the Physical Chemistry principles of Redox, Equilibria, Chemical Kinetics and Thermodynamics. The instruments used include Polarimeter, NMR Spectroscope, Electric Dipole measurement device, isotopic tracer, pH meter etc. CLASSIFICATION OF ORGANIC REACTIONS Organic Reactions in JEE syllabi can be classified as under : 1. Substitution reactions - (i) Free radical (ii) Nucleophilic-SN1, SN2,SNi (iii) Electrophilic 2. Elimination reactions-E1, E2 & E1cb 3. Addition reactions - (i) Electrophilic (ii) Nucleophilic (iii) Free Radical 4. Rearrangements The IITian’s Prashikshan Kendra Pvt. Ltd. Essential Notes on General Organic Chemistry (G.O.C.) 159 1. SUBSTITUTION REACTIONS A reaction in which one group or atom is replaced by another is called a substitution reaction. The incoming group is bonded to the same carbon to which the leaving group was bonded. -

17. Oxidation and Reduction Reactions 18
(11,12/94)(4,5/97)(02,3/07)(01/08) Neuman Chapter 17 Chapter 17 Oxidation and Reduction from Organic Chemistry by Robert C. Neuman, Jr. Professor of Chemistry, emeritus University of California, Riverside [email protected] <http://web.chem.ucsb.edu/~neuman/orgchembyneuman/> Chapter Outline of the Book ************************************************************************************** I. Foundations 1. Organic Molecules and Chemical Bonding 2. Alkanes and Cycloalkanes 3. Haloalkanes, Alcohols, Ethers, and Amines 4. Stereochemistry 5. Organic Spectrometry II. Reactions, Mechanisms, Multiple Bonds 6. Organic Reactions *(Not yet Posted) 7. Reactions of Haloalkanes, Alcohols, and Amines. Nucleophilic Substitution 8. Alkenes and Alkynes 9. Formation of Alkenes and Alkynes. Elimination Reactions 10. Alkenes and Alkynes. Addition Reactions 11. Free Radical Addition and Substitution Reactions III. Conjugation, Electronic Effects, Carbonyl Groups 12. Conjugated and Aromatic Molecules 13. Carbonyl Compounds. Ketones, Aldehydes, and Carboxylic Acids 14. Substituent Effects 15. Carbonyl Compounds. Esters, Amides, and Related Molecules IV. Carbonyl and Pericyclic Reactions and Mechanisms 16. Carbonyl Compounds. Addition and Substitution Reactions 17. Oxidation and Reduction Reactions 18. Reactions of Enolate Ions and Enols 19. Cyclization and Pericyclic Reactions *(Not yet Posted) V. Bioorganic Compounds 20. Carbohydrates 21. Lipids 22. Peptides, Proteins, and α−Amino Acids 23. Nucleic Acids ************************************************************************************** -

Mechanisms for Solvolytic Elimination and Substitution Reactions Involving Short-Lived Carbocation Intermediates
! " # $ % & # % '( )%*+",- .",- ** ,%/"$%*% 0*1%",% 00*1* 2332 ACTA UNIVERSITATIS UPSALIENSIS Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 743 Distributor: Uppsala University Library, Box 510, SE-751 20 Uppsala, Sweden Xiaofeng Zeng Mechanisms for Solvolytic Elimination and Substitution Reactions Involving Short-Lived Carbocation Intermediates Dissertation in Organic Chemistry to be publicly examined in The Svedberg Lecture Hall at the Institute of Chemistry, Uppsala University, on September 25, 2002 at 10.15 a.m., for the Degree of Doctor of Philosophy. The examination will be conducted in English. ABSTRACT Zeng X. 2002. Mechanisms for Solvolytic Elimination and Substitution Reactions Involving Short-Lived Carbocation Intermediates. Acta Univ. Ups., Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 743. 45 pp. Uppsala. ISBN 91-554-5389-9. Solvolysis reactions of a range of tertiary substrates in largely aqueous solvents were studied in such respects as E-deuterium kinetic isotope effects, linear free energy relationships and stereochemistry. Solvolysis of the fluorene derivatives 9-methyl–9-(2´-X-2´-propyl)fluorene (1-X, X = Cl, Br, OOCCF3) involves a very short-lived carbocation intermediate. The fraction of alkene is increased by addition of general bases, which can be expressed by a BrInsted parameter ȕ = 0.07. The kinetic deuterium isotope effects vary with solvent composition in a way which is not consistent with a common carbocation intermediate which has time to choose between dehydronation and addition of a solvent water molecule. In the absence of bases, the reaction of 4-chloro-4-(4´-nitrophenyl)pentan-2-one (2-Cl) proceeds through a short-lived carbocation intermediate yielding 4-(4´-nitrophenyl)-2-oxopent-4-ene (2-t-ne) as the main elimination product. -

Answers to Chapter 2 In-Chapter Problems
Answers To Chapter 2 In-Chapter Problems. 2.1. LDA is a strong base. Two E2 eliminations give an alkyne, which is deprotonated by the excess LDA to give an alkynyl anion. This species then reacts with MeI by an SN2 process. Cl OMe – HOMe N(i-Pr2) HH HOMe H OMe – H OMe – (i-Pr2)N N(i-Pr2) Me I MeO MeO Me 2.2(a). LDA deprotonates the C α to the ester, which adds to the aldehyde to give the aldol product after workup. O O O O O H3C – H3C N(i-Pr2) Et H C H H C 3 O 3 O CH3 CH3 O H C O 3 Et workup C H product H3 O O– CH3 2.2(b). BuLi deprotonates the C α to the nitrile, which adds to the ketone to give the aldol product after workup. O Et Et H O– Li Bu Et Et workup NC H NC H NC H product H H H 2.3. Make: C2–C3. Break: none. Note that because the NaCN is catalytic, its atoms are not incorporated into the product, and hence there is no need to number them. 1 O 3 O 1 O Ph cat. NaCN 2 H + 4 H 2 4 EtOH, H O Ph Ph 2 Ph 3 OH Chapter 2 2 C2 is electrophilic, and C4 is ... electrophilic! To make a bond between them, C2 must be turned into a nucleophile (umpolung). This must be the purpose of the –CN. Aldehydes are not acidic at the carbonyl C, so the –CN cannot simply deprotonate C2. -

4811 Et Et.Pdf
____________________________________________________________________________________________________ Subject Chemistry Paper No and Title Paper-9, Organic Chemistry-III (Reaction Mechanism-2) Module No and Title Module-7, Nucleophilic and Free Radical Addition Reactions of Alkenes Module Tag CHE_P9_M7 CHEMISTRY PAPER No. : Paper-9, Organic Chemistry-III (Reaction Mechanism-2) MODULE No. : Module-7, Nucleophilic and Free Radical Addition Reactions of Alkenes ____________________________________________________________________________________________________ TABLE OF CONTENTS 1. Learning Outcomes 2. Introduction 3. Nucleophilic Additions to Alkenes 3.1 Cyanoethylation 3.2 Hydro-cyano-addition 3.3 Michael Addition 3.4 Epoxidation of α,β-unsaturated Carbonyls 4. Free Radical Addition Reactions of Alkenes 4.1 Free radical addition of HBr 4.2 Free Radical Addition of Halomethanes 4.3 Other Free Radical Additions of Alkenes 6. Summary CHEMISTRY PAPER No. : Paper-9, Organic Chemistry-III (Reaction Mechanism-2) MODULE No. : Module-7, Nucleophilic and Free Radical Addition Reactions of Alkenes ____________________________________________________________________________________________________ 1. Learning Outcomes After studying this module, you shall be able to • Know about reactivity of alkene towards nucleophiles and free radicals • Learn about the alkene substrates that undergo nucleophilic additions • Familiarize with the mechanistic aspects of nucleophilic and free radical additions • Be acquainted with alkene’s nucleophilic and free radical additions of synthetic importance • Identify the stereochemistry and orientation of nucleophilic and free radical additions of alkenes 2. Introduction Besides the electrophilic addition reactions which constitute the major and most important of reactions of the carbon-cabon double bonds, alkenes also undergo few important nucleophilic additions and free radical addition reactions. Simple alkenes are not so much electrophilic that thay can undergo addition with nucleophiles.