Method 8141B
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Additive Interactions of Some Reduced-Risk Biocides and Two
www.nature.com/scientificreports OPEN Additive interactions of some reduced‑risk biocides and two entomopathogenic nematodes suggest implications for integrated control of Spodoptera litura (Lepidoptera: Noctuidae) Rashad Rasool Khan 1,2*, Muhammad Arshad 1*, Asad Aslam 3 & Muhammad Arshad 4 Higher volumes of conventional and novel chemical insecticides are applied by farmers to control resistant strains of armyworm (Spodoperta litura) in Pakistan without knowing their risks to the environment and to public health. Ten reduced‑risk insecticides were tested for their compatibility with two entomopathogenic nematodes (EPNs); Heterorhabditis indica and Steinernema carpocapsae rd against S. litura. The insecticide emamectin benzoate was highly toxic (LC50 = 2.97 mg/l) against 3 instar S. litura larvae when applied alone whereas, novaluron and methoxyfenozide were the least toxic (LC50 = 29.56 mg/l and 21.06 mg/l), respectively. All the insecticides proved harmless against the two EPNs even 96 h after treatment. Indoxacarb, fubendiamide and spinetoram produced the greatest mortalities (72–76%) of S. litura larvae after 72 h when applied in mixtures with H. indica. Lowest mortalities (44.00 ± 3.74% and 48.00 ± 2.89) were observed for mixtures of H. indica with methoxyfenozide and chlorfenapyr, respectively. The positive control treatments with both EPNs (S. carpocapsae and H. indica) produced > 50% mortality 96 h after treatment. For insecticide mixtures with S. carpocapsae, only indoxacarb produced 90% mortality of larvae, whereas, indoxacarb, fubendiamide, emamectin benzoate, and spinetoram produced 90–92% mortality of larvae when applied in mixtures with H. indica. Additive interactions (Chi‑square < 3.84) of EPN mixtures with reduced volumes of reduced‑risk insecticides suggest opportunities to develop more environmentally favorable pest management programs for S. -

Review of Azinphos-Methyl Was Undertaken by the Office of Chemical Safety (OCS), Which Considered All the Toxicological Data and Information Submitted for the Review
The reconsideration of the active constituent azinphos-methyl, registrations of products containing azinphos-methyl and approvals of their associated labels PRELIMINARY REVIEW FINDINGS Volume 1: Review Summary OCTOBER 2006 Canberra Australia Azinphos-methyl review – Preliminary Review Findings © Australian Pesticides & Veterinary Medicines Authority 2006 This work is copyright. Apart from any use permitted under the Copyright Act 1968, no part may be reproduced without permission from the Australian Pesticides & Veterinary Medicines Authority. The Australian Pesticides & Veterinary Medicines Authority publishes this preliminary review findings report for the active constituent azinphos-methyl and products containing azinphos- methyl. For further information about this review or the Pesticides Review Program, contact: Manager Chemical Review Australian Pesticides & Veterinary Medicines Authority PO Box E 240 KINGSTON ACT 2604 Australia Telephone: 61 2 6272 3213 Facsimile: 61 2 6272 3218 Email: [email protected] APVMA web site: http://www.apvma.gov.au i Azinphos-methyl review – Preliminary Review Findings FOREWORD The Australian Pesticides & Veterinary Medicines Authority (APVMA) is an independent statutory authority with responsibility for the regulation of agricultural and veterinary chemicals in Australia. Its statutory powers are provided in the Agvet Codes scheduled to the Agricultural and Veterinary Chemicals Code Act 1994. The APVMA can reconsider the approval of an active constituent, the registration of a chemical product or -

Malathion Human Health and Ecological Risk Assessment Final Report
SERA TR-052-02-02c Malathion Human Health and Ecological Risk Assessment Final Report Submitted to: Paul Mistretta, COR USDA/Forest Service, Southern Region 1720 Peachtree RD, NW Atlanta, Georgia 30309 USDA Forest Service Contract: AG-3187-C-06-0010 USDA Forest Order Number: AG-43ZP-D-06-0012 SERA Internal Task No. 52-02 Submitted by: Patrick R. Durkin Syracuse Environmental Research Associates, Inc. 5100 Highbridge St., 42C Fayetteville, New York 13066-0950 Fax: (315) 637-0445 E-Mail: [email protected] Home Page: www.sera-inc.com May 12, 2008 Table of Contents Table of Contents............................................................................................................................ ii List of Figures................................................................................................................................. v List of Tables ................................................................................................................................. vi List of Appendices ......................................................................................................................... vi List of Attachments........................................................................................................................ vi ACRONYMS, ABBREVIATIONS, AND SYMBOLS ............................................................... vii COMMON UNIT CONVERSIONS AND ABBREVIATIONS.................................................... x CONVERSION OF SCIENTIFIC NOTATION .......................................................................... -

The Analysis of Micro Amounts of Binapacryl, EPN, Methyl Parathion, and Parathion
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln U.S. Department of Agriculture: Agricultural Publications from USDA-ARS / UNL Faculty Research Service, Lincoln, Nebraska 12-1963 The Analysis of Micro Amounts of Binapacryl, EPN, Methyl Parathion, and Parathion Donald A. George USDA-ARS Follow this and additional works at: https://digitalcommons.unl.edu/usdaarsfacpub George, Donald A., "The Analysis of Micro Amounts of Binapacryl, EPN, Methyl Parathion, and Parathion" (1963). Publications from USDA-ARS / UNL Faculty. 1653. https://digitalcommons.unl.edu/usdaarsfacpub/1653 This Article is brought to you for free and open access by the U.S. Department of Agriculture: Agricultural Research Service, Lincoln, Nebraska at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Publications from USDA-ARS / UNL Faculty by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. George, Donald A. 0315·1 THE ANALYSIS OF rHeRO AHOUNTS OF BINAPl',CRYL, EPN, :tJlliTHYL PAR.ATHION, AND PAHATHION Journal of the A.O.A.~. (Vol. L6, No.6, 1963) The Analysis of Micro Amounts of Binapacryl~ EPN~ Methyl Parathion~ and Parathion By DONALD A. GEORGE (Entomology Research Division, Agricultural Research Service, U.S. Department of Agriculture, Yakima, Wash.) Aromatic nitrated insecticides can be cryl (2-sec-butyl-4,6-dinitrophenyl-3-methyl- determined by a microchemical method 2-butenoate) is determined by the method based on the Averell-Norris procednre. of Niagara Chemical Division of Food Ma The method is more sensitive than chinery Corp. (5). other known methods, and interference The microcolorimetric method presented from plant extracts is low. utilizes the reactions of the Averell-Norris procedure and has definite advantages over Aromatic nitrated insecticides have been the methods cited above. -

744 Hydrolysis of Chiral Organophosphorus Compounds By
[Frontiers in Bioscience, Landmark, 26, 744-770, Jan 1, 2021] Hydrolysis of chiral organophosphorus compounds by phosphotriesterases and mammalian paraoxonase-1 Antonio Monroy-Noyola1, Damianys Almenares-Lopez2, Eugenio Vilanova Gisbert3 1Laboratorio de Neuroproteccion, Facultad de Farmacia, Universidad Autonoma del Estado de Morelos, Morelos, Mexico, 2Division de Ciencias Basicas e Ingenierias, Universidad Popular de la Chontalpa, H. Cardenas, Tabasco, Mexico, 3Instituto de Bioingenieria, Universidad Miguel Hernandez, Elche, Alicante, Spain TABLE OF CONTENTS 1. Abstract 2. Introduction 2.1. Organophosphorus compounds (OPs) and their toxicity 2.2. Metabolism and treatment of OP intoxication 2.3. Chiral OPs 3. Stereoselective hydrolysis 3.1. Stereoselective hydrolysis determines the toxicity of chiral compounds 3.2. Hydrolysis of nerve agents by PTEs 3.2.1. Hydrolysis of V-type agents 3.3. PON1, a protein restricted in its ability to hydrolyze chiral OPs 3.4. Toxicity and stereoselective hydrolysis of OPs in animal tissues 3.4.1. The calcium-dependent stereoselective activity of OPs associated with PON1 3.4.2. Stereoselective hydrolysis commercial OPs pesticides by alloforms of PON1 Q192R 3.4.3. PON1, an enzyme that stereoselectively hydrolyzes OP nerve agents 3.4.4. PON1 recombinants and stereoselective hydrolysis of OP nerve agents 3.5. The activity of PTEs in birds 4. Conclusions 5. Acknowledgments 6. References 1. ABSTRACT Some organophosphorus compounds interaction of the racemic OPs with these B- (OPs), which are used in the manufacturing of esterases (AChE and NTE) and such interactions insecticides and nerve agents, are racemic mixtures have been studied in vivo, ex vivo and in vitro, using with at least one chiral center with a phosphorus stereoselective hydrolysis by A-esterases or atom. -

Parathion-Methyl
FAO SPECIFICATIONS AND EVALUATIONS FOR PLANT PROTECTION PRODUCTS PARATHION-METHYL O,O-dimethyl O-4-nitrophenyl phosphorothioate 2001 TABLE OF CONTENTS PARATHION-METHYL Page DISCLAIMER 3 INTRODUCTION 4 PART ONE 5 SPECIFICATIONS FOR PARATHION-METHYL PARATHION-METHYL INFORMATIONERROR! BOOKMARK NOT DEFINED. PARATHION-METHYL TECHNICAL MATERIAL 6 PARATHION-METHYL TECHNICAL CONCENTRATE 8 PARATHION-METHYL EMULSIFIABLE CONCENTRATE 10 PART TWO 13 2001 EVALUATION REPORT ON PARATHION-METHYL 14 Page 2 of 31 PARATHION-METHYL SPECIFICATIONS 2001 Disclaimer1 FAO specifications are developed with the basic objective of ensuring that pesticides complying with them are satisfactory for the purpose for which they are intended so that they may serve as an international point of reference. The specifications do not constitute an endorsement or warranty of the use of a particular pesticide for a particular purpose. Neither do they constitute a warranty that pesticides complying with these specifications are suitable for the control of any given pest, or for use in a particular area. Owing to the complexity of the problems involved, the suitability of pesticides for a particular application must be decided at the national or provincial level. Furthermore, the preparation and use of pesticides complying with these specifications are not exempted from any safety regulation or other legal or administrative provision applicable thereto. FAO shall not be liable for any injury, loss, damage or prejudice of any kind that may be suffered as a result of the preparation, transportation, sale or use of pesticides complying with these specifications. Additionally, FAO wishes to alert users of specifications to the fact that improper field mixing and/or application of pesticides can result in either a lowering or complete loss of efficacy. -
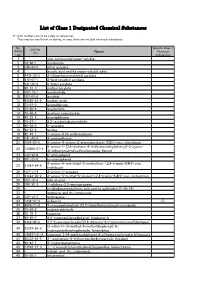
List of Class 1 Designated Chemical Substances
List of Class 1 Designated Chemical Substances *1:CAS numbers are to be solely as references. They may be insufficient or lacking, in case there are multiple chemical substances. No. Specific Class 1 CAS No. (PRTR Chemical (*1) Name Law) Substances 1 - zinc compounds(water-soluble) 2 79-06-1 acrylamide 3 140-88-5 ethyl acrylate 4 - acrylic acid and its water-soluble salts 5 2439-35-2 2-(dimethylamino)ethyl acrylate 6 818-61-1 2-hydroxyethyl acrylate 7 141-32-2 n-butyl acrylate 8 96-33-3 methyl acrylate 9 107-13-1 acrylonitrile 10 107-02-8 acrolein 11 26628-22-8 sodium azide 12 75-07-0 acetaldehyde 13 75-05-8 acetonitrile 14 75-86-5 acetone cyanohydrin 15 83-32-9 acenaphthene 16 78-67-1 2,2'-azobisisobutyronitrile 17 90-04-0 o-anisidine 18 62-53-3 aniline 19 82-45-1 1-amino-9,10-anthraquinone 20 141-43-5 2-aminoethanol 21 1698-60-8 5-amino-4-chloro-2-phenylpyridazin-3(2H)-one; chloridazon 5-amino-1-[2,6-dichloro-4-(trifluoromethyl)phenyl]-3-cyano- 22 120068-37-3 4[(trifluoromethyl)sulfinyl]pyrazole; fipronil 23 123-30-8 p-aminophenol 24 591-27-5 m-aminophenol 4-amino-6-tert-butyl-3-methylthio-1,2,4-triazin-5(4H)-one; 25 21087-64-9 metribuzin 26 107-11-9 3-amino-1-propene 27 41394-05-2 4-amino-3-methyl-6-phenyl-1,2,4-triazin-5(4H)-one; metamitron 28 107-18-6 allyl alcohol 29 106-92-3 1-allyloxy-2,3-epoxypropane 30 - n-alkylbenzenesulfonic acid and its salts(alkyl C=10-14) 31 - antimony and its compounds 32 120-12-7 anthracene 33 1332-21-4 asbestos ○ 34 4098-71-9 3-isocyanatomethyl-3,5,5-trimethylcyclohexyl isocyanate 35 78-84-2 isobutyraldehyde -

Environmental Health Criteria 145 Methyl Parathion
Environmental Health Criteria 145 Methyl parathion Please note that the layout and pagination of this web version are not identical with the printed version. Methyl parathion (EHC 145, 1992) INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY ENVIRONMENTAL HEALTH CRITERIA 145 METHYL PARATHION This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization First draft prepared by Dr R.F. Hertel and co-workers, Fraunhofer Institute of Toxicology and Aerosol Research, Hanover, Germany World Health Orgnization Geneva, 1993 The International Programme on Chemical Safety (IPCS) is a joint venture of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization. The main objective of the IPCS is to carry out and disseminate evaluations of the effects of chemicals on human health and the quality of the environment. Supporting activities include the development of epidemiological, experimental laboratory, and risk-assessment methods that could produce internationally comparable results, and the development of manpower in the field of toxicology. Other activities carried out by the IPCS include the development of know-how for coping with chemical accidents, -

Research and Special Programs Admin., DOT § 172.101
Research and Special Programs Admin., DOT § 172.101 TABLE 2 TO APPENDIX AÐRADIONUCLIDESÐ the class of the material must be determined Continued in accordance with § 173.2a of this sub- chapter. 3. This appendix contains two columns. (2)Ð (3)ÐReportable Atomic The first column, entitled ``S.M.P.'' (for se- (1)ÐRadionuclide Num- Quantity (RQ) ber Ci (TBq) vere marine pollutants), identifies whether a material is a severe marine pollutant. If the Ytterbium-169 .......................... 70 10 (.37) letters ``PP'' appear in this column for a ma- Ytterbium-175 .......................... 70 100 (3.7) terial, the material is a severe marine pol- Ytterbium-177 .......................... 70 1000 (37) lutant, otherwise it is not. The second col- Ytterbium-178 .......................... 70 1000 (37) umn, entitled ``Marine Pollutant'' , lists the Yttrium-86 ................................ 39 10 (.37) Yttrium-86m ............................. 39 1000 (37) marine pollutants. Yttrium-87 ................................ 39 10 (.37) 4. If a material not listed in this appendix Yttrium-88 ................................ 39 10 (.37) meets the criteria for a marine pollutant, as Yttrium-90 ................................ 39 10 (.37) provided in the General Introduction of the Yttrium-90m ............................. 39 100 (3.7) IMDG Code, Guidelines for the Identification Yttrium-91 ................................ 39 10 (.37) of Harmful Substances in Packaged Form, Yttrium-91m ............................. 39 1000 (37) Yttrium-92 ................................ 39 100 (3.7) the material may be transported as a marine Yttrium-93 ................................ 39 100 (3.7) pollutant in accordance with the applicable Yttrium-94 ................................ 39 1000 (37) requirements of this subchapter. Yttrium-95 ................................ 39 1000 (37) 5. If approved by the Associate Adminis- Zinc-62 ..................................... 30 100 (3.7) trator for Hazardous Materials Safety, a ma- Zinc-63 .................................... -

8 Toxicology of Organophosphate Nerve Agents
FYX FYX JWBK130-08 JWBK130-Marrs et al February 28, 2007 16:15 Char Count= 8 TOXICOLOGY OF ORGANOPHOSPHATE NERVE AGENTS Timothy C. Marrs Edentox Associates, Edenbridge, UK INTRODUCTION very high mammalian toxicity, were synthesized by Schrader in 1937 and a small pilot produc- The organophosphate (OP) anticholinesterases tion plant was set up at M¨unster-Lager. Later, include the chemical warfare nerve agents, a va- at D¨uhernfurt near Breslau in Prussian Silesia riety of OP pesticides (Ballantyne and Marrs, (now Bzerg Dolny and Wroclaw in Poland), a 1992) and, less well-known, a natural compound, production plant for these agents was established anatoxin-As (Dittmann and Wiegand, 2005). The and both tabun and sarin were manufactured in active ingredients of OP pesticides are used as quantity: in the case of tabun, 12 000 tonnes were drugs in human medicine, e.g. malathion to treat produced. Soman, another nerve agent, was also head-lice and metrifonate/trichlorfon in tropi- synthesized in Germany during the war, but only cal medicines. OPs are also used in veterinary manufactured in small quantities. Strangely, per- medicine notably as ectoparasiticides (Beesley, haps in view of the large stocks held by Ger- 1994). The chemical warfare nerve agents have a many, the nerve agents were not used in World much higher mammalian acute toxicity, particu- War II (UK Ministry of Defence, 1972) and ap- larly via the percutaneous and inhalation routes, pear to have been dumped in the Baltic sea. The than the OP pesticides. Additionally many chem- V agents similarly arose out of studies of puta- ical warfare agents are phosphonofluoridates and tive insecticides and VX has been variously re- phosphonothioates, while many pesticides are ported as being first synthesized at the Chemical = S type-phosphorothioates. -

EC84-1509 Field Crop Insect Control Guide for Nebraska Corn and Sorghum D
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Historical Materials from University of Nebraska- Extension Lincoln Extension 1984 EC84-1509 Field Crop Insect Control Guide for Nebraska Corn and Sorghum D. L. Keith L. L. Peters J. F. Witkowski Follow this and additional works at: http://digitalcommons.unl.edu/extensionhist Keith, D. L.; Peters, L. L.; and Witkowski, J. F., "EC84-1509 Field Crop Insect Control Guide for Nebraska Corn and Sorghum" (1984). Historical Materials from University of Nebraska-Lincoln Extension. 4433. http://digitalcommons.unl.edu/extensionhist/4433 This Article is brought to you for free and open access by the Extension at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Historical Materials from University of Nebraska-Lincoln Extension by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. ~I</ s EC 84-1509 85 67 FBf-/so 7 FIELD CROP INSECT CONTROL GUIDE FOR NEBRASKA c-L CORN AND SORGHUM D. L. Keith, L. L. Peters, J. F. Witkowski J. B. Campbell, A. F. Hagen Agricultural Extension Entomologists L. W. Andersen, IPM Specialist K. J. Jarvi, IPM Extension Assistant S. D. Danielson, Extension Technologist (Entomology) Insect control suggestions in this guide are based on University of Nebraska research results, U.S.D.A. recommendations and l abel registrations. Insect control is never perfect. The suggestions are designed to benefit Nebraska farmers when they need control programs. NebGuides containing additional information on identification, damaee and life cycles are listed under insect headings. They are available from county extension offices. Often the choice of a pesticide is based mainly on its cost. -

C:\SCANSOUTH\ATSDR\Wpd\Methyl Parathion.Wpd
TOXICOLOGICAL PROFILE FOR METHYL PARATHION U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service Agency for Toxic Substances and Disease Registry September 2001 METHYL PARATHION ii DISCLAIMER The use of company or product name(s) is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry. METHYL PARATHION iii UPDATE STATEMENT Toxicological profiles are revised and republished as necessary, but no less than once every three years. For information regarding the update status of previously released profiles, contact ATSDR at: Agency for Toxic Substances and Disease Registry Division of Toxicology/Toxicology Information Branch 1600 Clifton Road NE, E-29 Atlanta, Georgia 30333 METHYL PARATHION vi *Legislative Background The toxicological profiles are developed in response to the Superfund Amendments and Reauthorization Act (SARA) of 1986 (Public law 99-499) which amended the Comprehensive Environmental Response, Compensation, and Liability Act of 1980 (CERCLA or Superfund). This public law directed ATSDR to prepared toxicological profiles for hazardous substances most commonly found at facilities on the CERCLA National Priorities List and that pose the most significant potential threat to human health, as determined by ATSDR and the EPA. The availability of the revised priority list of 275 hazardous substances was announced in the Federal Register on November 17, 1997 (62 FR 61332). For prior versions of the list of substances, see Federal Register notices dated April 29, 1996 (61 FR 18744); April 17, 1987 (52 FR 12866); October 20, 1988 (53 FR 41280); October 26, 1989 (54 FR 43619); October 17, 1990 (55 FR 42067); October 17, 1991 (56 FR 52166); October 28, 1992 (57 FR 48801); and February 28, 1994 (59 FR 9486).