Technische Universtät München
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Spatiotemporal Evolution of Lakes Under Rapid Urbanization: a Case Study in Wuhan, China
water Article Spatiotemporal Evolution of Lakes under Rapid Urbanization: A Case Study in Wuhan, China Chao Wen 1, Qingming Zhan 1,* , De Zhan 2, Huang Zhao 2 and Chen Yang 3 1 School of Urban Design, Wuhan University, Wuhan 430072, China; [email protected] 2 China Construction Third Bureau Green Industry Investment Co., Ltd., Wuhan 430072, China; [email protected] (D.Z.); [email protected] (H.Z.) 3 College of Urban and Environmental Sciences, Peking University, Beijing 100871, China; [email protected] * Correspondence: [email protected]; Tel.: +86-139-956-686-39 Abstract: The impact of urbanization on lakes in the urban context has aroused continuous attention from the public. However, the long-term evolution of lakes in a certain megacity and the heterogeneity of the spatial relationship between related influencing factors and lake changes are rarely discussed. The evolution of 58 lakes in Wuhan, China from 1990 to 2019 was analyzed from three aspects of lake area, lake landscape, and lakefront ecology, respectively. The Multi-Scale Geographic Weighted Regression model (MGWR) was then used to analyze the impact of related influencing factors on lake area change. The investigation found that the total area of 58 lakes decreased by 15.3%. A worsening trend was found regarding lake landscape with the five landscape indexes of lakes dropping; in contrast, lakefront ecology saw a gradual recovery with variations in the remote sensing ecological index (RSEI) in the lakefront area. The MGWR regression results showed that, on the whole, the increase in Gross Domestic Product (GDP), RSEI in the lakefront area, precipitation, and humidity Citation: Wen, C.; Zhan, Q.; Zhan, contributed to lake restoration. -

Seasonal Succession of Bacterial Communities in Three Eutrophic Freshwater Lakes
International Journal of Environmental Research and Public Health Case Report Seasonal Succession of Bacterial Communities in Three Eutrophic Freshwater Lakes Bin Ji, Cheng Liu, Jiechao Liang and Jian Wang * Department of Water and Wastewater Engineering, Wuhan University of Science and Technology, Wuhan 430065, China; [email protected] (B.J.); [email protected] (C.L.); [email protected] (J.L.) * Correspondence: [email protected]; Tel.: +86-27-68893616 Abstract: Urban freshwater lakes play an indispensable role in maintaining the urban environment and are suffering great threats of eutrophication. Until now, little has been known about the seasonal bacterial communities of the surface water of adjacent freshwater urban lakes. This study reported the bacterial communities of three adjacent freshwater lakes (i.e., Tangxun Lake, Yezhi Lake and Nan Lake) during the alternation of seasons. Nan Lake had the best water quality among the three lakes as reflected by the bacterial eutrophic index (BEI), bacterial indicator (Luteolibacter) and functional prediction analysis. It was found that Alphaproteobacteria had the lowest abundance in summer and the highest abundance in winter. Bacteroidetes had the lowest abundance in winter, while Planctomycetes had the highest abundance in summer. N/P ratio appeared to have some relationships with eutrophication. Tangxun Lake and Nan Lake with higher average N/P ratios (e.g., N/P = 20) tended to have a higher BEI in summer at a water temperature of 27 ◦C, while Yezhi Lake with a relatively lower average N/P ratio (e.g., N/P = 14) tended to have a higher BEI in spring and autumn at a water temperature of 9–20 ◦C. -

Planning Strategy and Practice of Low-Carbon City Construction , 46 Th ISOCARP Congress 2010
Zhang Wentong, Planning Strategy and Practice of Low-carbon City Construction , 46 th ISOCARP Congress 2010 Planning Strategy and Practice of Low-carbon City Construction Development in Wuhan, China Zhang Wentong Yidong Hu I. Exploration on Planning of Low-carbon City Construction under the Global Context The concept of low-carbon is proposed in the context of responding to global climate change and advocating reducing the discharge of greenhouse gases in human’s production activities. While in the urban area, the low-carbon city is evolved gradually from the concept of ecological city, and these two can go hand in hand. The connotation of low-carbon city has also changed from the environment subject majoring in reducing carbon emission to a comprehensive subject including society, culture, economy and environment. Low-carbon city has become a macro-system synthesizing low-carbon technology, low-carbon production & consumption mode and mode of operation of low-carbon city. At last it will be amplified to the entire level of ecological city. The promotion of low-carbon city construction has a profound background of times and practical significance. Just as Professor Yu Li from Cardiff University of Great Britain has summed up, at least there are reasons from three aspects for the promotion of low-carbon city construction: firstly, reduce the emission of carbon through the building of ecological cities and return to a living style with the harmonious development between man and the nature; secondly, different countries hope to obtain a leading position in innovation through exploration on ecological city technology, idea and development mode and to lead the construction of sustainable city of the next generation; thirdly, to resolve the main problems in the country and local areas as well as the problem of “global warming”. -

Technical Assistance Consultant's Report People's Republic of China
Technical Assistance Consultant’s Report Project Number: 42011 November 2009 People’s Republic of China: Wuhan Urban Environmental Improvement Project Prepared by Easen International Co., Ltd in association with Kocks Consult GmbH For Wuhan Municipal Government This consultant’s report does not necessarily reflect the views of ADB or the Government concerned, and ADB and the Government cannot be held liable for its contents. (For project preparatory technical assistance: All the views expressed herein may not be incorporated into the proposed project’s design. ADB TA No. 7177- PRC Project Preparatory Technical Assistance WUHAN URBAN ENVIRONMENTAL IMPROVEMENT PROJECT Final Report November 2009 Volume I Project Analysis Consultant Executing Agency Easen International Co., Ltd. Wuhan Municipal Government in association with Kocks Consult GmbH ADB TA 7177-PRC Wuhan Urban Environmental Improvement Project Table of Contents WUHAN URBAN ENVIRONMENTAL IMPROVEMENT PROJECT ADB TA 7177-PRC FINAL REPORT VOLUME I PROJECT ANALYSIS TABLE OF CONTENTS Abbreviations Executive Summary Section 1 Introduction 1.1 Introduction 1-1 1.2 Objectives of the PPTA 1-1 1.3 Summary of Activities to Date 1-1 1.4 Implementation Arrangements 1-2 Section 2 Project Description 2.1 Project Rationale 2-1 2.2 Project Impact, Outcome and Benefits 2-2 2.3 Brief Description of the Project Components 2-3 2.4 Estimated Costs and Financial Plan 2-6 2.5 Synchronized ADB and Domestic Processes 2-6 Section 3 Technical Analysis 3.1 Introduction 3-1 3.2 Sludge Treatment and Disposal Component 3-1 3.3 Technical Analysis for Wuhan New Zone Lakes/Channels Rehabilitation, Sixin Pumping Station and Yangchun Lake Secondary Urban Center Lake Rehabilitation 3-51 3.4 Summary, Conclusions and Recommendations 3-108 Section 4 Environmental Impact Assessment 4.1 Status of EIAs and SEIA Approval 4-1 4.2 Overview of Chinese EIA Reports 4-1 Easen International Co. -

1 Opening Speech Time: 08:45 - 09:00 President’S Speech: Jian Guo Zhou, 10 Min Dean’S Speech: Baoshan Cui, 5 Min
Sponsored by National Key R&D Program of China (2018YFC1406400, 2016YFC0500402, 2017YFC0506603, 2018YFC0407403) Key Lab of Water and Sediment Science of the Ministry of Education (IAHR) International Association for Hydro-Environment Engineering and Research 1 Scientific Committee Name School Country John Bridgeman The University of Bradford U.K. Stuart Bunn Griffith University Australia John Cater The University of Auckland New Zealand Muk Chen Ong University of Stavanger Norway Taiwan, Jianzhong Chen Fooyin University China Carlo Gualtieri University of Naples Italy China Institute of Water Resources and Wei Huang China Hydropower Research Magnus Larson Lund University Sweden Yiping Li Hohai University China Pengzhi Lin Sichuan University China China Institute of Water Resources and Shu Liu China Hydropower Research William Nardin The University of Maryland U.S.A. Ling Qian Manchester Metropolitan University U.K. Benedict Rogers The University of Manchester U.K. Xinshan Song Donghua University China Marcel Stive Delft University of Technology Netherlands Guangzhi Sun Edith Cowan University Australia Xuelin Tang Chinese Agricultural University China Roger Wang Rutgers University U.S.A. Zheng Bing Wang Delft University of Technology Netherlands Yujun Yi Beijing Normal University China Xiao Yu University of Florida U.S.A. Zhonglong Zhang Portland State University U.S.A. 2 Conference Committee Chair: Jian Guo Zhou, Manchester Metropolitan University Co-Chairs: Baoshan Cui, Beijing Normal University Alistair G.L. Borthwick, The University -

Summary Environmental Impact Assessment Wuhan
SUMMARY ENVIRONMENTAL IMPACT ASSESSMENT WUHAN WASTEWATER MANAGEMENT PROJECT IN THE PEOPLE’S REPUBLIC OF CHINA July 2002 CURRENCY EQUIVALENTS (as of 15 June 2002) Currency Unit - yuan (CNY) CNY 1.00 = $0.121 $1.00 = CNY8.27 The exchange rate of the yuan is determined under a floating exchange rate system. In this report a rate of $1.00 = CNY8.27 is used. ABBREVIATIONS AAOV average annual output value A/C anaerobic plus modified carousel oxidation ditch process ADB Asian Development Bank A/O anaerobic/ oxic AP affected people BOD biochemical oxygen demand CODcr chemical oxygen demand EIA environmental impact assessment EIRR economic internal rate of return HPEPB Hubei Provincial Environmental Protection Bureau PPTA project preparatory technical assistance PRC People’s Republic of China PRO project resettlement office RAP Resettlement Action Plan RP Resettlement Plan SEPA State Environmental Protection Administration t/yr ton per year WMEPB Wuhan Municipal Environmental Protection Bureau WMWC Wuhan Municipal Wastewater Company WPMO Wuhan Project Management Office WWTP wastewater treatment plant WEIGHTS AND MEASURES ha Hectare km Kilometer km2 square kilometer m Meter m/s meter per second m3 cubic meter m3/day cubic meter per day t/yr ton per year NOTES: (i) The fiscal year (FY) of the government coincides with the calendar year. (ii) In this report, “$” refers to US dollars. CONTENTS MAP(s) ii I. INTRODUCTION 1 II. PROJECT DESCRIPTION 1 III. DESCRIPTION OF THE ENVIRONMENT 2 A. Topography and Geology 2 B. Climate and Rainfall 2 C. Hydrology 3 D. Ecological Resources 3 E. Water Quality and Pollution 4 F. Social and Economic Conditions 4 IV. -

Spatiotemporal Distribution and Mass Loadings of Perfluoroalkyl
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Institutional Repository of Guangzhou Institute of Geochemistry,CAS(GIGCAS OpenIR) Science of the Total Environment 493 (2014) 580–587 Contents lists available at ScienceDirect Science of the Total Environment journal homepage: www.elsevier.com/locate/scitotenv Spatiotemporal distribution and mass loadings of perfluoroalkyl substances in the Yangtze River of China Chang-Gui Pan, Guang-Guo Ying ⁎, Jian-Liang Zhao, You-Sheng Liu, Yu-Xia Jiang, Qian-Qian Zhang State Key Laboratory of Organic Geochemistry, Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, Guangzhou 510640, China HIGHLIGHTS • Contamination profiles of 18 PFCs in the Yangtze River investigated • PFOA was the predominant PFAS contaminant both in water and sediment. • The annual mass loading of total PFASs was 20900 kg/year. • Wuhan and Er'zhou contributed the most amounts of PFASs into the Yangtze River. • No significant seasonal variations observed for most PFASs in water. • Most PFAS concentrations were correlated positively with TN both in water and sediment. article info abstract Article history: A systematic investigation into contamination profiles of eighteen perfluoroalkyl substances (PFASs) in both sur- Received 13 March 2014 face water and sediments of Yangtze River was carried out by using high performance liquid chromatography– Received in revised form 9 June 2014 tandem mass spectrometry (HPLC–MS/MS) in summer and winter of 2013. The total concentrations of the Accepted 10 June 2014 PFASs in the water and sediment of Yangtze River ranged from 2.2 to 74.56 ng/L and 0.05 to 1.44 ng/g dry weights Available online 28 June 2014 (dw), respectively. -

A Case Study on the Lakes in Wuhan City, Hubei Province, China
International Journal of Environmental Research and Public Health Article A Fuzzy Comprehensive Assessment and Hierarchical Management System for Urban Lake Health: A Case Study on the Lakes in Wuhan City, Hubei Province, China Teng Wang 1,†, Jingjing Yan 2,†, Jinlong Ma 2, Fei Li 2,* , Chaoyang Liu 2, Ying Cai 2, Si Chen 3, Jingjing Zeng 2 and Yu Qi 2 1 Hubei Research Center of Water Affair, Hubei University of Economics, Wuhan 430073, China; [email protected] 2 School of Information and Safety Engineering, Zhongnan University of Economics and Law, Wuhan 430073, China; [email protected] (J.Y.); [email protected] (J.M.); [email protected] (C.L.); [email protected] (Y.C.); [email protected] (J.Z.); [email protected] (Y.Q.) 3 School of Resources and Environment, Hubei University, Wuhan 430062, China; [email protected] * Correspondence: [email protected] † These authors contributed equally to this work. Received: 13 October 2018; Accepted: 18 November 2018; Published: 22 November 2018 Abstract: Environmental assessment of eutrophication or heavy metals in urban lakes is an important reference for identifying the pollution degree and formulating pollution prevention strategies. At present, the most research on lake health states is often evaluated from a single angle for toxic metals pollution or eutrophication using the standard comparison method for both, the comprehensive trophic level index (TLI), and the health risk assessment for toxic metals. Moreover, the above deterministic methods probably lead to biased or unreliable assessment due to the randomness and fuzziness in environment system caused by natural change and human activities. -

Research Report of Urban Flood Risk Management Capacity | Ndrcc| Undp China
RESEARCH REPORT OF URBAN FLOOD RISK MANAGEMENT CAPACITY | NDRCC| UNDP CHINA RESEARCH REPORT OF URBAN FLOOD RISK MANAGEMENT CAPACITY THIS REPORT SUPPORT BY UNDP PROJECT 00083985 EMERGENCY MANAGEMENT AND COORDINATION SUPPORT IN CHINA THIS REPORT IS A COLLABORATION BETWEEN NATIONAL DISASTER REDUCTION CENTER OF CHINA AND UNITED NATIONS DEVELOPMENT PROGRAMME IN CHINA RESEARCH REPORT OF URBAN FLOOD RISK MANAGEMENT CAPACITY | NDRCC| UNDP CHINA Foreword The Research Report on Urban Flood Risk Management Capability was completed with the massive support of the United Nations Development Program (UNDP) in China and the National Disaster Reduction Center of China (NDRCC) affiliated to the Ministry of Civil Affairs. It is also the crystallization of joint efforts of all participating members. In recent years, the risk of urban flood disaster in China has become increasingly prominent, posing threats to not only the people's lives and property safety, but also the sustainable economic and social development. Against this backdrop, UNDP has established China Disaster Management and Coordination Support Project and commissioned the NDRCC to carry out the Research on Risk Management Capability of the Urban Flood Disaster. Based on field investigation, this Report analyzes the current situation, problems and deficiencies of urban flood disaster management in Wuhan and Ganzhou and puts forward some suggestions to strengthen the risk management of the urban flood disaster. The Report is divided into four chapters. The first chapter is the overview of flood disasters of China in 2016. The second and third chapters introduce the characteristics of flood disasters in Wuhan and Ganzhou by combining the city profiles and historical data of floods. -

Occurrence of Bothriocephalus Acheilognathi in Cyprinid Fish from Three Lakes in the Flood Plain of the Yangtze River, China
DISEASES OF AQUATIC ORGANISMS Vol. 41: 81-82,2000 Published May 25 Dis Aquat Org 1 l NOTE Occurrence of Bothriocephalus acheilognathi in cyprinid fish from three lakes in the flood plain of the Yangtze River, China P. Nie', G. T. Wang, W. J. Yao, Y. A. Zhang, Q. Gao State Key Laboratory of Freshwater Ecology and Biotechnology, and Laboratory of Fish Diseases, Institute of Hydrobiology, Chinese Academy of Sciences. Wuhan 430072. Hubei Province. PR China ABSTRACT. Cyprinid fish, Hemiculter leucisculus, Cultrich- A large number of lakes of various sizes are distrib- thys erythropterus and Culter dabryi, were sampled from uted in the flood plain of the Yangtze River (Chen & Liangzi, Honghu and Tangxun lakes in the flood plain of the Yangtze River. The cestode Bothriocephalus acheilognathi Xu 1995). In the present study, the 3 lakes sampled Yamaguti, 1934 was found in the 3 lakes, but C. erythropterus (Table 1) are located in Hubei province. All fish were sampled from Liangzi lake was found uninfected due proba- gill-netted, and transferred to the laboratory on the bly to the small sample size. Findings of the cestode in the 3 same day. The fork length and body weight of each lakes represent the first record of the parasite in the flood fish were measured. Intestines of the fish were plain of the Yangtze River, indicating that B. acheilognathi may be distributed much wider in China than previously rec- removed and examined under a microscope. Hel- ognized. minths found were identified alive and carmine- stained. KEY WORDS: Bothriocephalus acheilognathi . Hemiculter The cyprinids Hemiculter leucisculus (Basilewsky), leucisculus - Cultrichthys erythropterus Culter dabryi . -
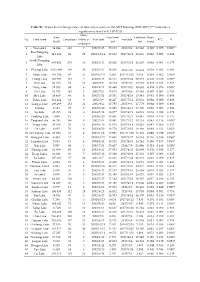
Table S1. Water Level Change Rates of Lakes/Reservoirs in the MLYB
Table S1. Water level change rates of lakes/reservoirs in the MLYB during 2002~2017 (“*” indicates a significance level of 0.1 (P<0.1)). Mean Area Start level End level Rates No Lake name Campaigns inliers of Start date End date R^2 P (km2) (m) (m) (m/y) campaigns 1 Wu Lake1 64.844 113 7 2002/7/15 25.372 2017/9/8 22.642 -0.097 0.075 0.003* East Dongting 2 841.366 83 25 2003/10/14 25.932 2017/12/4 22.939 0.026 0.002 0.684 Lake South Dongting 3 435.826 253 14 2002/6/13 28.920 2017/6/18 29.203 0.009 0.001 0.675 Lake 4 Poyang Lake 2835.484 319 38 2002/6/17 14.260 2017/7/8 18.938 0.019 0.002 0.403 5 Shijiu Lake 183.709 45 21 2003/10/31 5.982 2017/11/20 5.616 0.108 0.062 0.099* 6 Chang Lake 123.993 161 5 2002/6/13 32.111 2017/5/22 32.713 -0.081 0.126 0.000* 7 Wu Lake 20.166 66 10 2002/7/9 19.198 2017/9/6 18.783 -0.035 0.014 0.351 8 Dong Lake 32.503 88 6 2002/9/17 20.860 2017/10/5 20.685 -0.026 0.078 0.009* 9 Chi Lake 41.705 101 5 2002/7/6 15.074 2017/8/6 15.346 -0.007 0.001 0.793 10 Hou Lake 14.460 66 4 2002/7/31 18.551 2017/4/19 18.343 0.001 0.000 0.909 11 Futou Lake 101.414 92 9 2002/7/9 19.417 2017/7/12 20.033 0.004 0.000 0.889 12 Liangzi Lake 298.496 152 22 2002/9/4 17.787 2017/9/6 17.779 0.004 0.000 0.803 13 Ci Lake 9.126 75 5 2002/6/20 16.882 2016/4/13 17.320 0.006 0.006 0.524 14 Lu Lake 45.395 38 9 2004/5/18 18.877 2017/4/19 19.539 0.039 0.022 0.378 15 Yandong Lake 5.040 17 5 2004/6/20 18.907 2017/3/21 18.847 0.034 0.156 0.117 16 Tangxun Lake 44.783 146 8 2002/7/9 19.481 2017/7/12 19.733 0.045 0.116 0.000* 17 Wang Lake 45.109 23 11 2004/3/10 -

Lake Area Analysis Using Exponential Smoothing Model and Long Time-Series Landsat Images in Wuhan, China
sustainability Article Lake Area Analysis Using Exponential Smoothing Model and Long Time-Series Landsat Images in Wuhan, China Gonghao Duan 1,* ID and Ruiqing Niu 2 1 Faculty of Information Engineering, China University of Geosciences, Wuhan 430074, China 2 College of Geophysics and Geomatics, China University of Geosciences, Wuhan 430074, China; [email protected] * Correspondence: [email protected] Received: 14 November 2017; Accepted: 8 January 2018; Published: 9 January 2018 Abstract: The loss of lake area significantly influences the climate change in a region, and this loss represents a serious and unavoidable challenge to maintaining ecological sustainability under the circumstances of lakes that are being filled. Therefore, mapping and forecasting changes in the lake is critical for protecting the environment and mitigating ecological problems in the urban district. We created an accessible map displaying area changes for 82 lakes in the Wuhan city using remote sensing data in conjunction with visual interpretation by combining field data with Landsat 2/5/7/8 Thematic Mapper (TM) time-series images for the period 1987–2013. In addition, we applied a quadratic exponential smoothing model to forecast lake area changes in Wuhan city. The map provides, for the first time, estimates of lake development in Wuhan using data required for local-scale studies. The model predicted a lake area reduction of 18.494 km2 in 2015. The average error reached 0.23 with a correlation coefficient of 0.98, indicating that the model is reliable. The paper provided a numerical analysis and forecasting method to provide a better understanding of lake area changes.