Subject: Zavesca (Miglustat) [Gaucher Disease] Original Effective Date: 8/22/16
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

List of Covered Drugs
List of Covered Drugs Effective July 1, 2016 INTRODUCTION We are pleased to provide the 2011 Gold Coast Health Plan List of Covered Drugs as a useful reference and informational tool. The GCHP List of Covered Drugs can assist practitioners in selecting clinically appropriate and cost effective products for their patients. The information contained in the GCHP List of Covered Drugs is provided solely for the convenience of medical providers. We do not warrant or assure accuracy of such information nor is it intended to be comprehensive in nature. This List of Covered Drugs is not intended to be a substitute for the knowledge, expertise, skill and judgment of the medical provider in his or her choice of prescription drugs. All the information in this List of Covered Drugs is provided as a reference for drug therapy selection. Specific drug selection for an individual patient rests solely with the prescriber. We assume no responsibility for the actions or omissions of any medical provider based upon reliance, in whole or in part, on the information contained herein. The medical provider should consult the drug manufacturer's product literature or standard references for more detailed information. National guidelines can be found on the National Guideline Clearinghouse site at http://www.guideline.gov PREFACE The GCHP List of Covered Drugs is organized by sections. Each section is divided by therapeutic drug class primarily defined by mechanism of action. Unless exceptions are noted, generally all applicable dosage forms and strengths of the drug cited are included in the GCHP List of Covered Drugs. Generics should be considered the first line of prescribing. -

Intravenous Enzyme Replacement Therapy (ERT) for Gaucher Disease
UnitedHealthcare® Commercial Medical Benefit Drug Policy Intravenous Enzyme Replacement Therapy (ERT) for Gaucher Disease Policy Number: 2021D0048K Effective Date: April 1, 2021 Instructions for Use Table of Contents Page Related Commercial Policy Coverage Rationale ....................................................................... 1 • Provider Administered Drugs – Site of Care Applicable Codes .......................................................................... 3 Background.................................................................................... 3 Community Plan Policy Benefit Considerations .................................................................. 3 • Intravenous Enzyme Replacement Therapy (ERT) Clinical Evidence ........................................................................... 4 for Gaucher Disease U.S. Food and Drug Administration ............................................. 6 Centers for Medicare and Medicaid Services ............................. 6 References ..................................................................................... 7 Policy History/Revision Information ............................................. 8 Instructions for Use ....................................................................... 8 Coverage Rationale See Benefit Considerations This policy refers to the following drug products, all of which are intravenous enzyme replacement therapies used in the treatment of Gaucher disease: Cerezyme® (imiglucerase) Elelyso® (taliglucerase) VPRIV® (velaglucerase) -

Medical Policy Statement
MEDICAL POLICY STATEMENT Effective Next Annual Last Review / Date Review Date Revision Date 06/15/2011 06/15/2012 06/15/2011 Author Shelley Jones RN, CCM, Wendy Null RPh CSMG Medical Policy Statements are derived from literature based and supported clinical guidelines, nationally recognized utilization and technology assessment guidelines, other medical management industry standards, and published MCO clinical policy guidelines. Medically necessary services are those health care services or supplies which are proper and necessary for the diagnosis or treatment of disease, illness, or injury and without which the patient can be expected to suffer prolonged, increased or new morbidity, impairment of function, dysfunction of a body organ or part or significant pain and discomfort. These services meet the standards of good medical practice in the local area, are the lowest cost alternative and are not provided mainly for the convenience of the member or provider. A. SUBJECT Alglucerase (Ceredase) Infusion B. BACKGROUND Alglucerase (Ceredase) is a modified form of the enzyme, ß-glucocerebrosidase (ß-D- glucosyl-N-acylsphingosine glucohydrolase). Alglucerase (Ceredase) catalyzes the hydrolysis of the glycolipid, glucocerebroside, to glucose and ceramide as part of the normal degradation pathway for membrane lipids. Glucocerebroside is primarily derived from hematologic cell turnover. Gaucher disease is characterized by a functional deficiency in ß-glucocerebrosidase enzymatic activity and the resultant accumulation of lipid glucocerebroside in tissue macrophages, which become engorged and are termed Gaucher cells. The patient selection criteria outlined was derived from the FDA-approved prescribing information for alglucerase (Ceredase), the studies that were presented to the FDA in support of the pre-market approval application, and studies in the peer-reviewed published medical literature. -

Cerezyme, INN- Imiglucerase
SCIENTIFIC DISCUSSION This module reflects the initial scientific discussion for the approval of Cerezyme. This scientific discussion has been updated until 01 August 2003. For information on changes after this date please refer to module 8B. 1. Introduction Gaucher disease, also called glucosylceramide lipidosis or ß-glucocerebrosidase (GCR) deficiency, is the most common of the sphingolipidoses or lipid storage diseases, a group of diseases that are inherited in an autosomal recessive manner. It is characterised by the accumulation of glucocerebroside in tissue macrophages. Gaucher disease presents in three subtypes: type 1, the non- neuronopathic form; type 2, the acute neuronopathic form; and 3, the chronic neuronopathic form. Of these, type 1, non-neuronopathic form, is the most frequent and type 2, acute neuronopathic, is the least frequent. It has been estimated that type 1 Gaucher disease affects more than 20,000 patients world-wide. The clinical features of Gaucher disease include anaemia and thrombocytopenia due to splenic sequestration and bone marrow replacement by accumulating Gaucher cells. Splenomegaly and hepatomegaly are other frequent signs. Skeletal disease eventually leading to pain, stress fractures and osteonecrosis are common symptoms in type 1 and 3 Gaucher disease. The neuronopathic forms also present neurological abnormalities such as seizures, dementia, spasticity, ataxia and loss of intellectual functions. Enzyme replacement therapy (ERT) of Gaucher disease has been made feasible by the introduction of mannose-terminated ß-glucocerebrosidase. Mannose is a sugar, which is abundant on the surfaces of micro-organisms. It may be bound by the mannose receptor of macrophages. The mannose- termination of ß-glucocerebrosidase leads to a selective uptake of the enzyme by macrophages that are present in liver, spleen and skeleton. -

Vpriv-Pm-En.Pdf
PRODUCT MONOGRAPH INCLUDING PATIENT MEDICATION INFORMATION PrVPRIV® velaglucerase alfa Powder for Solution for Injection 400 U/vial, Intravenous Enzyme Replacement Therapy ATC code: A16AB10 Takeda Canada Inc. Date of Initial Approval: 22 Adelaide Street West, Suite 3800 October 1, 2010 Toronto Ontario M5H 4E3 Date of Revision: December 21, 2020 Submission Control No: 241844 VPRIV® and the VPRIV Logo® are registered trademarks of Shire Human Genetic Therapies, Inc. TAKEDA™ and the TAKEDA Logo® are trademarks of Takeda Pharmaceutical Company Limited, used under license. Page 1 of 24 RECENT MAJOR LABEL CHANGES Warnings and Precautions, Immunogenicity (8) 9/2020 Warnings and Precautions, Infusion-Related Reactions (8) 9/2020 Warnings and Precautions, Breast-feeding (8.1.2) 9/2020 TABLE OF CONTENTS PART I: HEALTH PROFESSIONAL INFORMATION ................................................................. 4 1 INDICATIONS .................................................................................................................4 2 CONTRAINDICATIONS ..................................................................................................4 4 DOSAGE AND ADMINISTRATION ................................................................................ 4 4.1 Dosing Considerations ..........................................................................................4 4.2 Recommended Dose and Dosage Adjustment ....................................................... 4 4.3 Administration .......................................................................................................4 -
![Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set](https://docslib.b-cdn.net/cover/8870/ehealth-dsi-ehdsi-v2-2-2-or-ehealth-dsi-master-value-set-1028870.webp)
Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set
MTC eHealth DSI [eHDSI v2.2.2-OR] eHealth DSI – Master Value Set Catalogue Responsible : eHDSI Solution Provider PublishDate : Wed Nov 08 16:16:10 CET 2017 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 1 of 490 MTC Table of Contents epSOSActiveIngredient 4 epSOSAdministrativeGender 148 epSOSAdverseEventType 149 epSOSAllergenNoDrugs 150 epSOSBloodGroup 155 epSOSBloodPressure 156 epSOSCodeNoMedication 157 epSOSCodeProb 158 epSOSConfidentiality 159 epSOSCountry 160 epSOSDisplayLabel 167 epSOSDocumentCode 170 epSOSDoseForm 171 epSOSHealthcareProfessionalRoles 184 epSOSIllnessesandDisorders 186 epSOSLanguage 448 epSOSMedicalDevices 458 epSOSNullFavor 461 epSOSPackage 462 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 2 of 490 MTC epSOSPersonalRelationship 464 epSOSPregnancyInformation 466 epSOSProcedures 467 epSOSReactionAllergy 470 epSOSResolutionOutcome 472 epSOSRoleClass 473 epSOSRouteofAdministration 474 epSOSSections 477 epSOSSeverity 478 epSOSSocialHistory 479 epSOSStatusCode 480 epSOSSubstitutionCode 481 epSOSTelecomAddress 482 epSOSTimingEvent 483 epSOSUnits 484 epSOSUnknownInformation 487 epSOSVaccine 488 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 3 of 490 MTC epSOSActiveIngredient epSOSActiveIngredient Value Set ID 1.3.6.1.4.1.12559.11.10.1.3.1.42.24 TRANSLATIONS Code System ID Code System Version Concept Code Description (FSN) 2.16.840.1.113883.6.73 2017-01 A ALIMENTARY TRACT AND METABOLISM 2.16.840.1.113883.6.73 2017-01 -
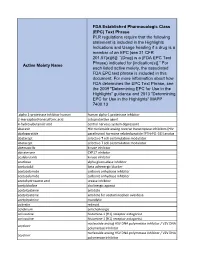
Active Moiety Name FDA Established Pharmacologic Class (EPC) Text
FDA Established Pharmacologic Class (EPC) Text Phrase PLR regulations require that the following statement is included in the Highlights Indications and Usage heading if a drug is a member of an EPC [see 21 CFR 201.57(a)(6)]: “(Drug) is a (FDA EPC Text Phrase) indicated for [indication(s)].” For Active Moiety Name each listed active moiety, the associated FDA EPC text phrase is included in this document. For more information about how FDA determines the EPC Text Phrase, see the 2009 "Determining EPC for Use in the Highlights" guidance and 2013 "Determining EPC for Use in the Highlights" MAPP 7400.13. .alpha. -

Estonian Statistics on Medicines 2016 1/41
Estonian Statistics on Medicines 2016 ATC code ATC group / Active substance (rout of admin.) Quantity sold Unit DDD Unit DDD/1000/ day A ALIMENTARY TRACT AND METABOLISM 167,8985 A01 STOMATOLOGICAL PREPARATIONS 0,0738 A01A STOMATOLOGICAL PREPARATIONS 0,0738 A01AB Antiinfectives and antiseptics for local oral treatment 0,0738 A01AB09 Miconazole (O) 7088 g 0,2 g 0,0738 A01AB12 Hexetidine (O) 1951200 ml A01AB81 Neomycin+ Benzocaine (dental) 30200 pieces A01AB82 Demeclocycline+ Triamcinolone (dental) 680 g A01AC Corticosteroids for local oral treatment A01AC81 Dexamethasone+ Thymol (dental) 3094 ml A01AD Other agents for local oral treatment A01AD80 Lidocaine+ Cetylpyridinium chloride (gingival) 227150 g A01AD81 Lidocaine+ Cetrimide (O) 30900 g A01AD82 Choline salicylate (O) 864720 pieces A01AD83 Lidocaine+ Chamomille extract (O) 370080 g A01AD90 Lidocaine+ Paraformaldehyde (dental) 405 g A02 DRUGS FOR ACID RELATED DISORDERS 47,1312 A02A ANTACIDS 1,0133 Combinations and complexes of aluminium, calcium and A02AD 1,0133 magnesium compounds A02AD81 Aluminium hydroxide+ Magnesium hydroxide (O) 811120 pieces 10 pieces 0,1689 A02AD81 Aluminium hydroxide+ Magnesium hydroxide (O) 3101974 ml 50 ml 0,1292 A02AD83 Calcium carbonate+ Magnesium carbonate (O) 3434232 pieces 10 pieces 0,7152 DRUGS FOR PEPTIC ULCER AND GASTRO- A02B 46,1179 OESOPHAGEAL REFLUX DISEASE (GORD) A02BA H2-receptor antagonists 2,3855 A02BA02 Ranitidine (O) 340327,5 g 0,3 g 2,3624 A02BA02 Ranitidine (P) 3318,25 g 0,3 g 0,0230 A02BC Proton pump inhibitors 43,7324 A02BC01 Omeprazole -

Miglustat (NB-DNJ)
Article pubs.acs.org/molecularpharmaceutics Therapeutic Strategies for Gaucher Disease: Miglustat (NB-DNJ) as a Pharmacological Chaperone for Glucocerebrosidase and the Different Thermostability of Velaglucerase Alfa and Imiglucerase Olga Abian,*, ,!,§,% Pilar Alfonso,*, ,!,¥ Adrian Velazquez-Campoy,§,#,∇ Pilar Giraldo, ,!,¥,Ë Miguel Pocovi,!,¥,# and Javier Sancho§,# Unidad de Investigación Traslacional, Miguel Servet Universitary Hospital, Zaragoza, Spain !Aragon Health Sciences Institute (I+CS), Zaragoza, Spain §Institute of Biocomputation and Physics of Complex Systems (BIFI), University of Zaragoza, BIFI-IQFR (CSIC)-Joint Unit, Spain %Centro de Investigación Biomedicá en Red en el Área Tematicá de Enfermedades Hepaticaś y Digestivas (CIBERehd) and ¥Centro de Investigación en Red de Enfermedades Raras (CIBERER), ISCIII, Spain #Department of Biochemistry and Cellular and Molecular Biology, University of Zaragoza, Spain ∇Fundación ARAID (ARAID-BIFI), Diputación General de Aragón, Spain ËService of Hematology, Miguel Servet Universitary Hospital, Zaragoza, Spain ABSTRACT: Gaucher disease (GD) is a disorder of glycosphingolipid metabolism caused by deficiency of lysosomal glucocerebrosidase (GlcCerase) activity, due to conformationally or functionally defective variants, resulting in progressive deposition of glycosylceramide in macrophages. The glucose analogue, N-butyldeoxynojirimycin (NB- DNJ, miglustat), is an inhibitor of the ceramide-specific glycosyltransfer- ase, which catalyzes the first step of glycosphingolipid biosynthesis -

Long-Term Follow-Up and Sudden Unexpected Death in Gaucher Disease Type 3 in Egypt
Long-term follow-up and sudden unexpected death in Gaucher disease type 3 in Egypt Magy Abdelwahab, MD, ABSTRACT PhD Objective: To describe the long-term follow-up and distinct phenotype of a large cohort of patients Derek Blankenship, PhD with Gaucher disease type 3 on enzyme replacement therapy (ERT) in Egypt. Raphael Schiffmann, Methods: A prospective cohort study of 78 patients on ERT who were followed for up to 9 years MD, MHSc with yearly evaluations that included EEG and cognitive testing. Results: Of the patients, 73% were homozygous for the L444P GBA1 mutation; all but 7 were Correspondence to neurologically symptomatic. Supranuclear gaze palsy with variable but stable cognitive function Dr. Schiffmann: was present in 91% of patients. Convergent strabismus and bulbar dysfunction were noted in Raphael.schiffmann@ baylorhealth.edu or 22% and 37%, respectively. Features of oppositional defiant disorder were present in 54% of Dr. Abdelwahab: patients. Twenty-three patients (30%) developed seizures while on ERT for 1–9 years. Of those, [email protected] 12 patients (15%) died suddenly and unexpectedly at a mean age of 6.7 6 5.0 years (range 1.5– 18). Sudden death was usually associated with a seizure disorder or a terminal seizure, but 7 of 12 patients had a preceding normal EEG. An additional 11% had background slowing or epilep- togenic activity on EEG without clinical seizures. There were 3 familial cases of sudden unex- pected death. Conclusions: Despite having the most common GBA1 genotype known to be associated with neuronopathic Gaucher disease, patients with Gaucher disease type 3 in Egypt have a phenotype and a clinical outcome on ERT that are very different from those observed in other populations. -

Quantum of Effectiveness Evidence in FDA's Approval of Orphan Drugs
Quantum of Effectiveness Evidence in FDA’s Approval of Orphan Drugs Cataloguing FDA’s Flexibility in Regulating Therapies for Persons with Rare Disorders by Frank J. Sasinowski, M.S., M.P.H., J.D.1 Chairman of the Board National Organization for Rare Disorders One of the key underlying issues facing the development of effective for their intended uses. all drugs, and particularly orphan drugs, is what kind of evi- dence the Food and Drug Administration (FDA) requires for FDA has for many decades acknowledged that there is a need approval. The Federal Food, Drug, and Cosmetic [FD&C] Act for flexibility in applying its standard for approval. For ex- provides that for FDA to grant approval for a new drug, there ample, one of FDA’s regulations states that: “FDA will ap- must be “substantial evidence” of effectiveness derived from prove an application after it determines that the drug meets the “adequate and well-controlled investigations.” This language, statutory standards for safety and effectiveness… While the which dates from 1962, provides leeway for FDA medical re- statutory standards apply to all drugs, the many kinds of drugs viewers to make judgments as to what constitutes “substantial that are subject to the statutory standards and the wide range evidence” of a drug’s effectiveness, that is, of its benefit to of uses for those drugs demand flexibility in applying the stan- patients. dards. Thus FDA is required to exercise its scientific judgment to determine the kind and quantity of data and information an The sole law that applies specifically to orphan drugs, the Or- applicant is required to provide for a particular drug to meet phan Drug Act of 1983, provided financial incentives for drug the statutory standards.” 21 C.F.R. -

Intravenous Enzyme Replacement Therapy (ERT) for Gaucher Disease
UnitedHealthcare® Oxford Clinical Policy Intravenous Enzyme Replacement Therapy (ERT) for Gaucher Disease Policy Number: PHARMACY 270.16 T2 Effective Date: April 1, 2021 Instructions for Use Table of Contents Page Related Policies Coverage Rationale ....................................................................... 1 • Acquired Rare Disease Drug Therapy Exception Prior Authorization Requirements ................................................ 3 Process Applicable Codes .......................................................................... 3 • Drug Coverage Guidelines Background ................................................................................... 3 • Experimental/Investigational Treatment Benefit Considerations .................................................................. 4 • Experimental/Investigational Treatment for NJ Plans Clinical Evidence ........................................................................... 4 U.S. Food and Drug Administration ............................................. 6 • Provider Administered Drugs – Site of Care References ..................................................................................... 7 Policy History/Revision Information ............................................. 8 Instructions for Use ....................................................................... 8 Coverage Rationale See Benefit Considerations This policy refers to the following drug products, all of which are intravenous enzyme replacement therapies used in the treatment of Gaucher disease: